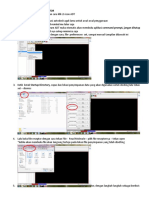
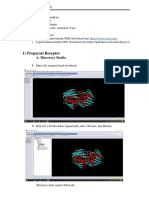
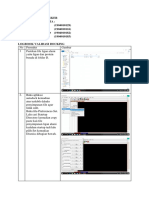
Anda mungkin juga menyukai
- Tutorial Docking Autodock4.2 (New)Dokumen3 halamanTutorial Docking Autodock4.2 (New)MochamadFathurrBelum ada peringkat
- B2 1 1908010096 Khairunnisa Lapres p2Dokumen14 halamanB2 1 1908010096 Khairunnisa Lapres p2Khairun nisaBelum ada peringkat
- Tutorial Docking Andi BasoDokumen7 halamanTutorial Docking Andi BasoHIDAYATBelum ada peringkat
- Tutorial Molecular Docking Dengan Autodock VinaDokumen7 halamanTutorial Molecular Docking Dengan Autodock Vinaludya pulungBelum ada peringkat
- Laporan Praktikum Molecular DockingDokumen14 halamanLaporan Praktikum Molecular DockingIsnainiBelum ada peringkat
- Panduan Molecular Docking Autodock 4.0Dokumen17 halamanPanduan Molecular Docking Autodock 4.0alisia difianaBelum ada peringkat
- Tutorial AutodockDokumen11 halamanTutorial AutodockFina RepitiyanaBelum ada peringkat
- Cara Docking Senyawa Dengan Autodock 4Dokumen10 halamanCara Docking Senyawa Dengan Autodock 4Carlos RumangunBelum ada peringkat
- B2 - 3 - 1908010108 - Muhammad Naufal Rabbani - P2Dokumen14 halamanB2 - 3 - 1908010108 - Muhammad Naufal Rabbani - P2siti salma haniyyahBelum ada peringkat
- Tutorial AutodockDokumen11 halamanTutorial AutodockA.S.H projecBelum ada peringkat
- Mari Belajar Molecular DockingDokumen5 halamanMari Belajar Molecular DockingAnggie WindrianiBelum ada peringkat
- Modul Biokimia Proyek 2: Molecular DockingDokumen31 halamanModul Biokimia Proyek 2: Molecular DockingNISRINA NAJLA HUWAIDABelum ada peringkat
- Alur DockingDokumen1 halamanAlur DockingAnnisa Zesika DewiBelum ada peringkat
- DOCKING FEBYANTI SAFITRI Dan GUSTI MAHARINIDokumen7 halamanDOCKING FEBYANTI SAFITRI Dan GUSTI MAHARININasrullah HamdaniBelum ada peringkat
- Molecular Docking PlantsDokumen21 halamanMolecular Docking Plantswiwin winingsihBelum ada peringkat
- Molecular DockingDokumen6 halamanMolecular DockingYasaKaryadaBelum ada peringkat
- Cake Rna RasiyyDokumen5 halamanCake Rna RasiyyEfanBelum ada peringkat
- Kimia MedisinalDokumen19 halamanKimia MedisinalAnjali WidhiyaniBelum ada peringkat
- Additional ModulDokumen14 halamanAdditional ModulYudi PratamaBelum ada peringkat
- Laporan DockingDokumen14 halamanLaporan DockingMarytta Indah Heryanti100% (1)
- Modul Bioinfor DockingDokumen17 halamanModul Bioinfor DockinggitaBelum ada peringkat
- Laporan Praktikum Kimia KomputasiDokumen19 halamanLaporan Praktikum Kimia KomputasiSHELFI RENITABelum ada peringkat
- Kimed 6Dokumen7 halamanKimed 6rizaldi rahmatullahBelum ada peringkat
- Laporan Praktikum DockingDokumen14 halamanLaporan Praktikum DockingMuhammadRezaPahleviBelum ada peringkat
- Logbook Ligan Alami Dan Reseptor Antikanker-DikonversiDokumen10 halamanLogbook Ligan Alami Dan Reseptor Antikanker-DikonversiNamira NaaziahBelum ada peringkat
- Log Book Preparasi Ligan Dan Reseptor - Kelompok Antihistamin - Farmasi D 2019Dokumen10 halamanLog Book Preparasi Ligan Dan Reseptor - Kelompok Antihistamin - Farmasi D 2019Namira NaaziahBelum ada peringkat
- DOCKINGDokumen8 halamanDOCKINGNovita ekaBelum ada peringkat
- Tutorial Docking AutodockDokumen3 halamanTutorial Docking AutodockFuzi Fauzia Fajrin Fauzia FajrinBelum ada peringkat
- Laporan Kimkom Modul 5 Fix Alsa Sah AlhamdulilahDokumen19 halamanLaporan Kimkom Modul 5 Fix Alsa Sah AlhamdulilahAlsa giani MaheshaBelum ada peringkat
- KELOMPOK ANTIBAKTERI-ligan Dan Reseptor-Farmasi 3dDokumen9 halamanKELOMPOK ANTIBAKTERI-ligan Dan Reseptor-Farmasi 3delicenrlBelum ada peringkat
- Modul SFBDokumen4 halamanModul SFBAtika afritamaBelum ada peringkat
- Carakerja TyyDokumen2 halamanCarakerja TyyEfanBelum ada peringkat
- Logbook Kimed IiDokumen11 halamanLogbook Kimed IiTatang SupriatnaBelum ada peringkat
- Tutorial Simulasi Molecular Docking Menggunakan DOCK6Dokumen6 halamanTutorial Simulasi Molecular Docking Menggunakan DOCK6putryaBelum ada peringkat
- Turorial Molecular DockingDokumen34 halamanTurorial Molecular DockinggedeBelum ada peringkat
- LaporanDokumen13 halamanLaporanLia NurhayatiBelum ada peringkat
- Molecular DockingDokumen57 halamanMolecular DockingpritaultimardianBelum ada peringkat
- Kelompok 3 - Shift D - Molecular Docking of Curcumin With Cox 2 ReceptorDokumen17 halamanKelompok 3 - Shift D - Molecular Docking of Curcumin With Cox 2 ReceptorPradita Rizki IrianiBelum ada peringkat
- Laporan Praktikum Kimia Komputasi ANALISIS DAN VALIDASI PENAMBATAN MOLEKUL (MOLECULAR DOCKING)Dokumen11 halamanLaporan Praktikum Kimia Komputasi ANALISIS DAN VALIDASI PENAMBATAN MOLEKUL (MOLECULAR DOCKING)Gita Namira Masri0% (1)
- Panduan DockingDokumen19 halamanPanduan DockingRetno Agus PratiwiBelum ada peringkat
- Tutorial Autodock VinaDokumen22 halamanTutorial Autodock VinapmuftiaBelum ada peringkat
- DokingDokumen29 halamanDokingAnjali WidhiyaniBelum ada peringkat
- LOGBOOK KELOMPOK ANTIKANKER VALIDASI DOCKING-dikonversiDokumen12 halamanLOGBOOK KELOMPOK ANTIKANKER VALIDASI DOCKING-dikonversiNamira NaaziahBelum ada peringkat
- Bab Iii Metode Penelitian A. Desain Penelitian: in Silico Molecular Docking PLANTSDokumen9 halamanBab Iii Metode Penelitian A. Desain Penelitian: in Silico Molecular Docking PLANTSNiken SwBelum ada peringkat
- Pembuatan Reseptor Dalam PDBQT Dengan Autodock ToolDokumen4 halamanPembuatan Reseptor Dalam PDBQT Dengan Autodock Toolsalsabila JacobBelum ada peringkat
- Kimkim HendrikDokumen11 halamanKimkim HendrikHendrik SeptiadiBelum ada peringkat
- Logbook Docking Senyawa Ligan Alami, Ligan Induk, Ligan Turunan Dan Senyawa Obat - Kelampok 6 Farmasi D 2019Dokumen10 halamanLogbook Docking Senyawa Ligan Alami, Ligan Induk, Ligan Turunan Dan Senyawa Obat - Kelampok 6 Farmasi D 2019Elis Nurul IkhlasBelum ada peringkat
- Cinta Skripsi Banget-46-54Dokumen9 halamanCinta Skripsi Banget-46-54Fyan VergarraBelum ada peringkat
- DatabaseDokumen38 halamanDatabaseHezekiel HBelum ada peringkat
- Bioinformatika 4Dokumen9 halamanBioinformatika 4pwd bambangBelum ada peringkat
- Bioinformatika StepsDokumen8 halamanBioinformatika StepsAriane BeninaBelum ada peringkat
- Cara Mendownload Data Klorofil Dan Suhu Permukaan LautDokumen5 halamanCara Mendownload Data Klorofil Dan Suhu Permukaan LautSITI NURHOLISAHBelum ada peringkat
- Tutorial Autodock 4.2 - 2018Dokumen6 halamanTutorial Autodock 4.2 - 2018lokawati muliaBelum ada peringkat
- Modul Sendiri LolDokumen33 halamanModul Sendiri LolyaminBelum ada peringkat
- Laporan Praktikum Kimia Komputasi Virtual Screening-DikonversiDokumen17 halamanLaporan Praktikum Kimia Komputasi Virtual Screening-DikonversiTiktok SerBelum ada peringkat
- Anasya Ridha N. - Molecular DockingDokumen16 halamanAnasya Ridha N. - Molecular DockingvinamalauBelum ada peringkat
- Laporan Kimkom Modul 4Dokumen19 halamanLaporan Kimkom Modul 4Alsa giani MaheshaBelum ada peringkat
- Modifikasi Senyawa KalkonDokumen4 halamanModifikasi Senyawa KalkonAyyu WidyazmaraBelum ada peringkat
- Bioinformatika 4 - An Nissa Ade Astary Dewi - 119180020 - 2021Dokumen6 halamanBioinformatika 4 - An Nissa Ade Astary Dewi - 119180020 - 2021annissa 119180020Belum ada peringkat
- Form-QA-01-004-01 Formulir IV Otorisasi Penanganan Penyimpangan Rev 1Dokumen2 halamanForm-QA-01-004-01 Formulir IV Otorisasi Penanganan Penyimpangan Rev 1syifa nurul ainiBelum ada peringkat
- Soal Responsi Praktikum SterilDokumen5 halamanSoal Responsi Praktikum Sterilsyifa nurul ainiBelum ada peringkat
- Terpenoid IDokumen7 halamanTerpenoid ILea AfrianaBelum ada peringkat
- Orde Reaksi Waktu Paruh PDFDokumen30 halamanOrde Reaksi Waktu Paruh PDFTheresia Iga AyuBelum ada peringkat
- Orde Reaksi Waktu Paruh PDFDokumen30 halamanOrde Reaksi Waktu Paruh PDFTheresia Iga AyuBelum ada peringkat
- Soal Pengantar Farmasi Tipe A PDFDokumen6 halamanSoal Pengantar Farmasi Tipe A PDFsyifa nurul ainiBelum ada peringkat
- Soal Kalkulus DiferensialDokumen3 halamanSoal Kalkulus Diferensialsyifa nurul ainiBelum ada peringkat
- Soal Pengantar Farmasi Tipe ADokumen6 halamanSoal Pengantar Farmasi Tipe Asyifa nurul ainiBelum ada peringkat
- Soal Prakt. Mikrobiologi Dan VirologiDokumen6 halamanSoal Prakt. Mikrobiologi Dan Virologisyifa nurul ainiBelum ada peringkat
- Soal Aplikasi Sistem KomputerDokumen6 halamanSoal Aplikasi Sistem Komputersyifa nurul aini100% (1)
- Soal Mikrobiologi Dan VirologiDokumen5 halamanSoal Mikrobiologi Dan Virologisyifa nurul ainiBelum ada peringkat
- FitokimiaDokumen3 halamanFitokimiasyifa nurul ainiBelum ada peringkat
- Bioteknologi Farmasi Tipe ADokumen6 halamanBioteknologi Farmasi Tipe Asyifa nurul aini100% (1)
- MFI II Tipe A EditDokumen3 halamanMFI II Tipe A Editsyifa nurul ainiBelum ada peringkat
- Presentasi Aik 5Dokumen14 halamanPresentasi Aik 5syifa nurul ainiBelum ada peringkat
- Kolinergik FixDokumen22 halamanKolinergik Fixsyifa nurul ainiBelum ada peringkat
- BAB II SalinaDokumen7 halamanBAB II Salinasyifa nurul ainiBelum ada peringkat
- Kolinergik FixDokumen22 halamanKolinergik Fixsyifa nurul ainiBelum ada peringkat
- Jurnal Bawang PutihDokumen117 halamanJurnal Bawang Putihsyifa nurul ainiBelum ada peringkat
- Tuga Kimed Kolinergik FixDokumen22 halamanTuga Kimed Kolinergik Fixsyifa nurul aini100% (1)
- Tuga Kimed Kolinergik FixDokumen22 halamanTuga Kimed Kolinergik Fixsyifa nurul aini0% (1)
- Bab I BSLTDokumen3 halamanBab I BSLTsyifa nurul ainiBelum ada peringkat
- RPKPS Manajemen Pemasaran FarmasiDokumen6 halamanRPKPS Manajemen Pemasaran Farmasisyifa nurul ainiBelum ada peringkat
- Hubungan Stuktur, Sifat Kimia Dengan Proses AbsorpsiDokumen43 halamanHubungan Stuktur, Sifat Kimia Dengan Proses Absorpsisyifa nurul ainiBelum ada peringkat
- Pendahuluan Kimia MedisinalDokumen5 halamanPendahuluan Kimia Medisinalsyifa nurul ainiBelum ada peringkat
- 1 Slide Manajemen Pemasaran FarmasiDokumen40 halaman1 Slide Manajemen Pemasaran Farmasivivipanda68Belum ada peringkat
- Kuliah UUF Ke-2 Baru HAM, Per-UU &Dokumen40 halamanKuliah UUF Ke-2 Baru HAM, Per-UU &syifa nurul ainiBelum ada peringkat
- Kajian RisikoDokumen29 halamanKajian Risikosyifa nurul ainiBelum ada peringkat
- Kuliah UUF Ke-1 Baru PIHDokumen33 halamanKuliah UUF Ke-1 Baru PIHsyifa nurul ainiBelum ada peringkat