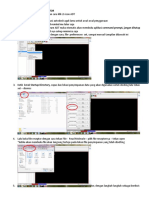
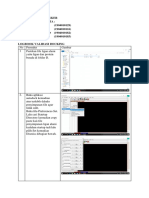
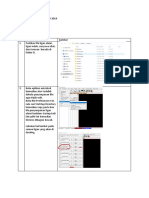
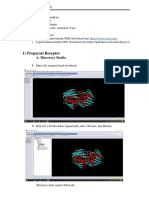
Anda mungkin juga menyukai
- Laporan Praktikum Kimia KomputasiDokumen19 halamanLaporan Praktikum Kimia KomputasiSHELFI RENITABelum ada peringkat
- Turorial Molecular DockingDokumen34 halamanTurorial Molecular DockinggedeBelum ada peringkat
- Panduan Molecular Docking Autodock 4.0Dokumen17 halamanPanduan Molecular Docking Autodock 4.0alisia difianaBelum ada peringkat
- Modul Biokimia Proyek 2: Molecular DockingDokumen31 halamanModul Biokimia Proyek 2: Molecular DockingNISRINA NAJLA HUWAIDABelum ada peringkat
- Tutorial AutodockDokumen11 halamanTutorial AutodockFina RepitiyanaBelum ada peringkat
- Tutorial AutodockDokumen11 halamanTutorial AutodockA.S.H projecBelum ada peringkat
- Kimia Komputasi Modul 5Dokumen23 halamanKimia Komputasi Modul 5rillynBelum ada peringkat
- Mari Belajar Molecular DockingDokumen5 halamanMari Belajar Molecular DockingAnggie WindrianiBelum ada peringkat
- LOGBOOK KELOMPOK ANTIKANKER VALIDASI DOCKING-dikonversiDokumen12 halamanLOGBOOK KELOMPOK ANTIKANKER VALIDASI DOCKING-dikonversiNamira NaaziahBelum ada peringkat
- Kimed 6Dokumen7 halamanKimed 6rizaldi rahmatullahBelum ada peringkat
- Laporan Kimkom Modul 5 Fix Alsa Sah AlhamdulilahDokumen19 halamanLaporan Kimkom Modul 5 Fix Alsa Sah AlhamdulilahAlsa giani MaheshaBelum ada peringkat
- Bioinformatika StepsDokumen8 halamanBioinformatika StepsAriane BeninaBelum ada peringkat
- Tutorial Autodock 4.2 - 2018Dokumen6 halamanTutorial Autodock 4.2 - 2018lokawati muliaBelum ada peringkat
- Logbook Docking Senyawa Ligan Alami, Ligan Induk, Ligan Turunan Dan Senyawa Obat - Kelampok 6 Farmasi D 2019Dokumen10 halamanLogbook Docking Senyawa Ligan Alami, Ligan Induk, Ligan Turunan Dan Senyawa Obat - Kelampok 6 Farmasi D 2019Elis Nurul IkhlasBelum ada peringkat
- B2 1 1908010096 Khairunnisa Lapres p2Dokumen14 halamanB2 1 1908010096 Khairunnisa Lapres p2Khairun nisaBelum ada peringkat
- Tutorial Docking AutodockDokumen3 halamanTutorial Docking AutodockFuzi Fauzia Fajrin Fauzia FajrinBelum ada peringkat
- Tutorial Molecular Docking Dengan Autodock VinaDokumen7 halamanTutorial Molecular Docking Dengan Autodock Vinaludya pulungBelum ada peringkat
- Molecular DockingDokumen6 halamanMolecular DockingYasaKaryadaBelum ada peringkat
- Logbook Kimed IiDokumen11 halamanLogbook Kimed IiTatang SupriatnaBelum ada peringkat
- Tutorial Docking Autodock4.2 (New)Dokumen3 halamanTutorial Docking Autodock4.2 (New)MochamadFathurrBelum ada peringkat
- Tutorial Autodock VinaDokumen22 halamanTutorial Autodock VinapmuftiaBelum ada peringkat
- Laporan Praktikum Molecular DockingDokumen14 halamanLaporan Praktikum Molecular DockingIsnainiBelum ada peringkat
- Cara Docking Senyawa Dengan Autodock 4Dokumen10 halamanCara Docking Senyawa Dengan Autodock 4Carlos RumangunBelum ada peringkat
- DOCKINGDokumen8 halamanDOCKINGNovita ekaBelum ada peringkat
- Tutorial Docking Andi BasoDokumen7 halamanTutorial Docking Andi BasoHIDAYATBelum ada peringkat
- Modul Sendiri LolDokumen33 halamanModul Sendiri LolyaminBelum ada peringkat
- 11-BRIYAN NUGRAHA PUTRA-06-Praktek Pencarian Dan Pengurutan DataDokumen16 halaman11-BRIYAN NUGRAHA PUTRA-06-Praktek Pencarian Dan Pengurutan DataGofur SincanBelum ada peringkat
- Carakerja TyyDokumen2 halamanCarakerja TyyEfanBelum ada peringkat
- BAB IV - Nurul AeniDokumen4 halamanBAB IV - Nurul AeniKudus Abdul azizBelum ada peringkat
- Modul Bioinfor DockingDokumen17 halamanModul Bioinfor DockinggitaBelum ada peringkat
- 06-ALFANDI PRIYATNA-06-Praktek Pencarian Dan Pengurutan DataDokumen15 halaman06-ALFANDI PRIYATNA-06-Praktek Pencarian Dan Pengurutan DataGofur SincanBelum ada peringkat
- Bioinformatika 4Dokumen9 halamanBioinformatika 4pwd bambangBelum ada peringkat
- Tutorial Simulasi Molecular Docking Menggunakan DOCK6Dokumen6 halamanTutorial Simulasi Molecular Docking Menggunakan DOCK6putryaBelum ada peringkat
- Additional ModulDokumen14 halamanAdditional ModulYudi PratamaBelum ada peringkat
- Contoh AplikasiDokumen17 halamanContoh AplikasiAiguille DevoirBelum ada peringkat
- Validasi ReseptorDokumen3 halamanValidasi Reseptorsyifa nurul ainiBelum ada peringkat
- Modul 7 (Penambatan Molekul)Dokumen2 halamanModul 7 (Penambatan Molekul)Ruth AnnekeBelum ada peringkat
- Protokol Pelatihan BioinformatikaDokumen21 halamanProtokol Pelatihan BioinformatikaI EBelum ada peringkat
- B2 - 3 - 1908010108 - Muhammad Naufal Rabbani - P2Dokumen14 halamanB2 - 3 - 1908010108 - Muhammad Naufal Rabbani - P2siti salma haniyyahBelum ada peringkat
- Cinta Skripsi Banget-46-54Dokumen9 halamanCinta Skripsi Banget-46-54Fyan VergarraBelum ada peringkat
- Laporan Praktikum Modul 2 Sistem DigitalDokumen8 halamanLaporan Praktikum Modul 2 Sistem Digitalkyrgios50% (2)
- Laporan Praktikum Kimia Komputasi Virtual Screening-DikonversiDokumen17 halamanLaporan Praktikum Kimia Komputasi Virtual Screening-DikonversiTiktok SerBelum ada peringkat
- DOCKING FEBYANTI SAFITRI Dan GUSTI MAHARINIDokumen7 halamanDOCKING FEBYANTI SAFITRI Dan GUSTI MAHARININasrullah HamdaniBelum ada peringkat
- Laporan Praktikum DockingDokumen14 halamanLaporan Praktikum DockingMuhammadRezaPahleviBelum ada peringkat
- Acara 3. Desain PrimerDokumen10 halamanAcara 3. Desain PrimerSalsabila Fitra RamadantiBelum ada peringkat
- Septiani Nurcahyanti - 24020221140090 - Lapres Acara 4 BioinformatikaDokumen14 halamanSeptiani Nurcahyanti - 24020221140090 - Lapres Acara 4 BioinformatikaSeptiani NurcahyantiBelum ada peringkat
- Trie Oktaviani - Penambatan Molekul (Molecular Docking)Dokumen17 halamanTrie Oktaviani - Penambatan Molekul (Molecular Docking)Trie OktavianiBelum ada peringkat
- Bab 3Dokumen6 halamanBab 3sukmawati Farm017Belum ada peringkat
- Combinational Logic SequenceDokumen9 halamanCombinational Logic SequenceYogi Salomo Mangontang PratamaBelum ada peringkat
- Perencanaan Transportasi Operatif - Solusi Dalam Rantai Pasokan Barang Konsumen-Physica-1-1Dokumen40 halamanPerencanaan Transportasi Operatif - Solusi Dalam Rantai Pasokan Barang Konsumen-Physica-1-1La Ode Agustian NurrahmanBelum ada peringkat
- Cake Rna RasiyyDokumen5 halamanCake Rna RasiyyEfanBelum ada peringkat
- Kimkim HendrikDokumen11 halamanKimkim HendrikHendrik SeptiadiBelum ada peringkat
- Resume 6Dokumen9 halamanResume 6004Nelly JuentiBelum ada peringkat
- Pre Test TaqiyuddinDokumen3 halamanPre Test TaqiyuddinAhmad TaqiyuddinBelum ada peringkat
- Nelly Juenti - Resume 6 - Kelompok 2Dokumen9 halamanNelly Juenti - Resume 6 - Kelompok 2004Nelly JuentiBelum ada peringkat
- Pengolahan Dinsar Dengan Software Snap-1Dokumen27 halamanPengolahan Dinsar Dengan Software Snap-1An Nisa Tri Rahmawati50% (2)
- Qiime2 ProtocolDokumen11 halamanQiime2 ProtocolfarrassBelum ada peringkat
- Object Detection Tensorflow API Part 2 Data PreparationDokumen4 halamanObject Detection Tensorflow API Part 2 Data PreparationBaiqunyBelum ada peringkat
- Molecular DockingDokumen57 halamanMolecular DockingpritaultimardianBelum ada peringkat
- Filsafat Sejarah-1Dokumen4 halamanFilsafat Sejarah-1Lia NurhayatiBelum ada peringkat
- Sejarah Pergerakan Nasional (Jaman Jepang)Dokumen27 halamanSejarah Pergerakan Nasional (Jaman Jepang)Lia NurhayatiBelum ada peringkat
- ProdiaDokumen3 halamanProdiaLia NurhayatiBelum ada peringkat
- Definisi IDokumen9 halamanDefinisi ILia NurhayatiBelum ada peringkat