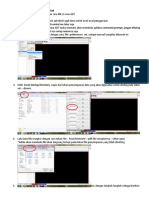
Anda mungkin juga menyukai
- Laporan Kimkom Modul 5 Fix Alsa Sah AlhamdulilahDokumen19 halamanLaporan Kimkom Modul 5 Fix Alsa Sah AlhamdulilahAlsa giani MaheshaBelum ada peringkat
- LaporanDokumen13 halamanLaporanLia NurhayatiBelum ada peringkat
- Panduan Molecular Docking Autodock 4.0Dokumen17 halamanPanduan Molecular Docking Autodock 4.0alisia difianaBelum ada peringkat
- B2 1 1908010096 Khairunnisa Lapres p2Dokumen14 halamanB2 1 1908010096 Khairunnisa Lapres p2Khairun nisaBelum ada peringkat
- B2 - 3 - 1908010108 - Muhammad Naufal Rabbani - P2Dokumen14 halamanB2 - 3 - 1908010108 - Muhammad Naufal Rabbani - P2siti salma haniyyahBelum ada peringkat
- Turorial Molecular DockingDokumen34 halamanTurorial Molecular DockinggedeBelum ada peringkat
- Modul Biokimia Proyek 2: Molecular DockingDokumen31 halamanModul Biokimia Proyek 2: Molecular DockingNISRINA NAJLA HUWAIDABelum ada peringkat
- Mari Belajar Molecular DockingDokumen5 halamanMari Belajar Molecular DockingAnggie WindrianiBelum ada peringkat
- Kimed 6Dokumen7 halamanKimed 6rizaldi rahmatullahBelum ada peringkat
- Carakerja TyyDokumen2 halamanCarakerja TyyEfanBelum ada peringkat
- Laporan Praktikum Molecular DockingDokumen14 halamanLaporan Praktikum Molecular DockingIsnainiBelum ada peringkat
- Cinta Skripsi Banget-46-54Dokumen9 halamanCinta Skripsi Banget-46-54Fyan VergarraBelum ada peringkat
- Kimkim HendrikDokumen11 halamanKimkim HendrikHendrik SeptiadiBelum ada peringkat
- Molecular DockingDokumen6 halamanMolecular DockingYasaKaryadaBelum ada peringkat
- Protokol Pelatihan BioinformatikaDokumen21 halamanProtokol Pelatihan BioinformatikaI EBelum ada peringkat
- Laporan Praktikum Kimia Komputasi Virtual Screening-DikonversiDokumen17 halamanLaporan Praktikum Kimia Komputasi Virtual Screening-DikonversiTiktok SerBelum ada peringkat
- Laporan Praktikum DockingDokumen14 halamanLaporan Praktikum DockingMuhammadRezaPahleviBelum ada peringkat
- Trie Oktaviani - Penambatan Molekul (Molecular Docking)Dokumen17 halamanTrie Oktaviani - Penambatan Molekul (Molecular Docking)Trie OktavianiBelum ada peringkat
- Modul 7 (Penambatan Molekul)Dokumen2 halamanModul 7 (Penambatan Molekul)Ruth AnnekeBelum ada peringkat
- Tutorial AutodockDokumen11 halamanTutorial AutodockFina RepitiyanaBelum ada peringkat
- Jurnal Laporan Praktikum Kimia MedisinalDokumen4 halamanJurnal Laporan Praktikum Kimia MedisinalOllive Filsa HawaBelum ada peringkat
- Tutorial AutodockDokumen11 halamanTutorial AutodockA.S.H projecBelum ada peringkat
- Kimia Komputasi Modul 5Dokumen23 halamanKimia Komputasi Modul 5rillynBelum ada peringkat
- Tutorial Autodock 4.2 - 2018Dokumen6 halamanTutorial Autodock 4.2 - 2018lokawati muliaBelum ada peringkat
- Tutorial Molecular Docking Dengan Autodock VinaDokumen7 halamanTutorial Molecular Docking Dengan Autodock Vinaludya pulungBelum ada peringkat
- Anasya Ridha N. - Molecular DockingDokumen16 halamanAnasya Ridha N. - Molecular DockingvinamalauBelum ada peringkat
- DOCKINGDokumen8 halamanDOCKINGNovita ekaBelum ada peringkat
- Cake Rna RasiyyDokumen5 halamanCake Rna RasiyyEfanBelum ada peringkat
- Maulidina Athadi Gayo - Molecular DockingDokumen11 halamanMaulidina Athadi Gayo - Molecular DockingMaulidina100% (3)
- Tutorial Docking AutodockDokumen3 halamanTutorial Docking AutodockFuzi Fauzia Fajrin Fauzia FajrinBelum ada peringkat
- Jurnal Praktikum Kimia Medisinal Rezalia PDFDokumen4 halamanJurnal Praktikum Kimia Medisinal Rezalia PDFRezalia PutriBelum ada peringkat
- Tutorial Docking Andi BasoDokumen7 halamanTutorial Docking Andi BasoHIDAYATBelum ada peringkat
- Tutorial Simulasi Molecular Docking Menggunakan DOCK6Dokumen6 halamanTutorial Simulasi Molecular Docking Menggunakan DOCK6putryaBelum ada peringkat
- Laporan Kimkom Modul 4Dokumen19 halamanLaporan Kimkom Modul 4Alsa giani MaheshaBelum ada peringkat
- Laporan Praktikum Kimia Komputasi ANALISIS DAN VALIDASI PENAMBATAN MOLEKUL (MOLECULAR DOCKING)Dokumen11 halamanLaporan Praktikum Kimia Komputasi ANALISIS DAN VALIDASI PENAMBATAN MOLEKUL (MOLECULAR DOCKING)Gita Namira Masri0% (1)
- DOckingDokumen10 halamanDOckingLindayenBelum ada peringkat
- Bab 3Dokumen6 halamanBab 3sukmawati Farm017Belum ada peringkat
- Cara Docking Senyawa Dengan Autodock 4Dokumen10 halamanCara Docking Senyawa Dengan Autodock 4Carlos RumangunBelum ada peringkat
- Tutorial Docking Autodock4.2 (New)Dokumen3 halamanTutorial Docking Autodock4.2 (New)MochamadFathurrBelum ada peringkat
- Analisis Docking MolekulerDokumen9 halamanAnalisis Docking Molekulerdesi ratna permatasariBelum ada peringkat
- Laporan DockingDokumen14 halamanLaporan DockingMarytta Indah Heryanti100% (1)
- IndahDokumen37 halamanIndahIndah Siti HardiyantiBelum ada peringkat
- Modifikasi Senyawa KalkonDokumen4 halamanModifikasi Senyawa KalkonAyyu WidyazmaraBelum ada peringkat
- Additional ModulDokumen14 halamanAdditional ModulYudi PratamaBelum ada peringkat
- Kelompok 3 Yunisa Anugrahwati G84130040 Laporan 7Dokumen12 halamanKelompok 3 Yunisa Anugrahwati G84130040 Laporan 7YunisaBelum ada peringkat
- Digdo 9178 Bioinfo Pak SyaifurDokumen22 halamanDigdo 9178 Bioinfo Pak SyaifurDigdo SudigyoBelum ada peringkat
- Tutorial Autodock VinaDokumen22 halamanTutorial Autodock VinapmuftiaBelum ada peringkat
- Laporan Praktikum Kimia Medisinal 4Dokumen21 halamanLaporan Praktikum Kimia Medisinal 4Aldi Yustian100% (5)
- Soal Post-Test TPACK 1Dokumen4 halamanSoal Post-Test TPACK 1Andini SalsabilaBelum ada peringkat
- Molecular DockingDokumen72 halamanMolecular DockingToksikologi D4Belum ada peringkat
- 06-ALFANDI PRIYATNA-06-Praktek Pencarian Dan Pengurutan DataDokumen15 halaman06-ALFANDI PRIYATNA-06-Praktek Pencarian Dan Pengurutan DataGofur SincanBelum ada peringkat
- Laporan Praktikum Kimia Komputasi Virtual ScreeningDokumen8 halamanLaporan Praktikum Kimia Komputasi Virtual ScreeningYouth BlueBelum ada peringkat
- Modul Praktikum KimedDokumen34 halamanModul Praktikum KimedVincentBelum ada peringkat
- Validasi ReseptorDokumen3 halamanValidasi Reseptorsyifa nurul ainiBelum ada peringkat
- Logbook Kimed IiDokumen11 halamanLogbook Kimed IiTatang SupriatnaBelum ada peringkat
- Kelompok 3 - Shift D - Molecular Docking of Curcumin With Cox 2 ReceptorDokumen17 halamanKelompok 3 - Shift D - Molecular Docking of Curcumin With Cox 2 ReceptorPradita Rizki IrianiBelum ada peringkat
- LKPD MK PPM Kelompok 6 - Entalpi ReaksiDokumen23 halamanLKPD MK PPM Kelompok 6 - Entalpi Reaksi21BSefti Regita Putri SikloheksanolBelum ada peringkat
- Bioinformatika 4Dokumen9 halamanBioinformatika 4pwd bambangBelum ada peringkat
- Molecular DockingDokumen57 halamanMolecular DockingpritaultimardianBelum ada peringkat