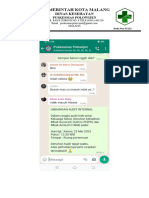
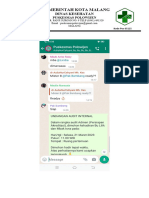
Anda mungkin juga menyukai
- CEDE KEPALADokumen20 halamanCEDE KEPALAEndo100% (3)
- KEJANG PASKA STROKEDokumen20 halamanKEJANG PASKA STROKEAnry Umar0% (2)
- Status EpileptikusDokumen28 halamanStatus EpileptikusSiti Sholihatun NisaBelum ada peringkat
- GMO Pada Stroke IskemikDokumen22 halamanGMO Pada Stroke IskemikandreBelum ada peringkat
- Gejala Kejang Akut Yang Diakibatkan Oleh Gangguan ElektrolitDokumen13 halamanGejala Kejang Akut Yang Diakibatkan Oleh Gangguan ElektrolitMuhammad Nur Syaiful MohidinBelum ada peringkat
- Patofisiologi Epilepsi Pasca StrokeDokumen4 halamanPatofisiologi Epilepsi Pasca StrokeBayu Firdaus SiradzBelum ada peringkat
- Laporan Pendahuluan Status EpileptikusDokumen27 halamanLaporan Pendahuluan Status EpileptikusTri Khusniyatu; MaromiBelum ada peringkat
- Tinpusss EpilepsiDokumen9 halamanTinpusss EpilepsiAnnisa Ena RBelum ada peringkat
- Gejala Kejang Akut Yang Diakibatkan Oleh Gangguan ElektrolitDokumen14 halamanGejala Kejang Akut Yang Diakibatkan Oleh Gangguan ElektrolitFransiska OktavianiBelum ada peringkat
- Diagnosis Banding ST EpiDokumen3 halamanDiagnosis Banding ST EpiHary SaktiawanBelum ada peringkat
- Kejang Pasca StrokeDokumen14 halamanKejang Pasca StrokeKiki Kiko0% (2)
- STATUS EPILEPTIKUSDokumen13 halamanSTATUS EPILEPTIKUSPatricia Renata100% (1)
- Pato MeisyaDokumen5 halamanPato MeisyaYasmin RaudhatulBelum ada peringkat
- LAPORAN PENDAHULUAN KejangDokumen26 halamanLAPORAN PENDAHULUAN KejangIndi meiliaBelum ada peringkat
- Epilepsi Gangguan Elektrolit BaruuDokumen35 halamanEpilepsi Gangguan Elektrolit BaruuAgung Anom Arie WiradanaBelum ada peringkat
- Laporan Pendahuluan Status EpileptikusDokumen28 halamanLaporan Pendahuluan Status EpileptikusHusna Ardiana0% (3)
- Graciela M. Pandeirot - Askep EpilepsiDokumen46 halamanGraciela M. Pandeirot - Askep EpilepsiIndri Sudirman IlyasBelum ada peringkat
- Laporan Pendahuluan Status EpileptikusDokumen26 halamanLaporan Pendahuluan Status EpileptikusLida Nurlainah nurlainahBelum ada peringkat
- LP EpilepsiDokumen26 halamanLP EpilepsiRisa HartatiBelum ada peringkat
- LP EpilepsiDokumen14 halamanLP Epilepsiega zinniaBelum ada peringkat
- Laporan Pendahuluan EpilepsiDokumen8 halamanLaporan Pendahuluan EpilepsiMan DiazBelum ada peringkat
- Neurointensive CareDokumen24 halamanNeurointensive CaretamictrBelum ada peringkat
- LP - KMB - Epilepsi AndiDokumen25 halamanLP - KMB - Epilepsi AndiAndi PangestuBelum ada peringkat
- Status Epileptikus AnakDokumen18 halamanStatus Epileptikus AnakHayatul UlfaBelum ada peringkat
- Status EpileptikusDokumen27 halamanStatus EpileptikusMutiaOktavianiBelum ada peringkat
- Askep EpilepsiDokumen34 halamanAskep EpilepsiAlwan AlfazariBelum ada peringkat
- LP Cidera Kepala, Kasus, Sop, JurnalDokumen44 halamanLP Cidera Kepala, Kasus, Sop, JurnalNabila alfaishaBelum ada peringkat
- Kejang Dan GGN ElektrolitDokumen22 halamanKejang Dan GGN ElektrolitIwing Dwi PurwandiBelum ada peringkat
- Status Epileptikus KomplikasiDokumen1 halamanStatus Epileptikus KomplikasiHary SaktiawanBelum ada peringkat
- Laporan PendahuluanDokumen25 halamanLaporan Pendahuluandwirabiatul adwiyahaliBelum ada peringkat
- Gangguan Tidur pada EpilepsiDokumen41 halamanGangguan Tidur pada EpilepsiAnnaJuntakBelum ada peringkat
- Referat Status EpileptikusDokumen18 halamanReferat Status EpileptikushanhyunbinBelum ada peringkat
- LP EpilepsiDokumen21 halamanLP Epilepsierva nidaBelum ada peringkat
- PATOFISIOLOGI EPILEPSIDokumen11 halamanPATOFISIOLOGI EPILEPSIMYusufEffendiBelum ada peringkat
- Manajemen Kedaruratan NeurologiDokumen34 halamanManajemen Kedaruratan NeurologiDhinie NovianiBelum ada peringkat
- Loket LoketDokumen18 halamanLoket LoketNdah MelizaBelum ada peringkat
- EPILEPSIDokumen15 halamanEPILEPSIirafenaraniBelum ada peringkat
- EpilepsiDokumen23 halamanEpilepsiHelpa Puspasari Part IIIBelum ada peringkat
- Epilepsi Ika SaraswatiDokumen15 halamanEpilepsi Ika SaraswatiIkaa SaraswatiiBelum ada peringkat
- Laporan Pendahuluan EpilepsiDokumen22 halamanLaporan Pendahuluan EpilepsiDeby BulagaBelum ada peringkat
- LP Epilepsi OkDokumen23 halamanLP Epilepsi OkAgung LarashatiBelum ada peringkat
- Lpkas (3) - 2Dokumen37 halamanLpkas (3) - 2Ninda AfriniBelum ada peringkat
- DETEKSI DINI EPILEPSIDokumen21 halamanDETEKSI DINI EPILEPSIOtneil K. KeliatBelum ada peringkat
- EPILEPSI BaruDokumen22 halamanEPILEPSI Baruabigail_ndmBelum ada peringkat
- LP - PICU - Epilepsi - Ajeng NijarDokumen26 halamanLP - PICU - Epilepsi - Ajeng NijarEva NurjanahBelum ada peringkat
- Lpkas (3) - 3Dokumen37 halamanLpkas (3) - 3Ninda AfriniBelum ada peringkat
- Laporan EpilepsiDokumen17 halamanLaporan EpilepsiRina SofianingrumBelum ada peringkat
- LP EpilepsiDokumen19 halamanLP EpilepsiALFIAN ARDIANUSBelum ada peringkat
- Tinjauan PustakaDokumen13 halamanTinjauan PustakatoonBelum ada peringkat
- LP Epilepsi MultazamDokumen13 halamanLP Epilepsi MultazamUsna UlandariBelum ada peringkat
- Patofisiologi dan Pengobatan Stroke: Status Saat Ini dan Perspektif Masa DepanDokumen2 halamanPatofisiologi dan Pengobatan Stroke: Status Saat Ini dan Perspektif Masa DepanNabilahBelum ada peringkat
- Bab I Syok FixxxxDokumen9 halamanBab I Syok FixxxxWiwit Indriyani AslinaBelum ada peringkat
- Epilepsi Laporan dan AsuhanDokumen18 halamanEpilepsi Laporan dan AsuhangetrudisBelum ada peringkat
- Laporan Hasil KegiatanDokumen5 halamanLaporan Hasil KegiatanRainbow DashieBelum ada peringkat
- Notulen Audit PIS-PKDokumen5 halamanNotulen Audit PIS-PKRainbow DashieBelum ada peringkat
- DUPAKDokumen5 halamanDUPAKRainbow DashieBelum ada peringkat
- Bu Afidah Dinkes Jatim Peran Dokter Dalam Menurunkan AKI AKBDokumen9 halamanBu Afidah Dinkes Jatim Peran Dokter Dalam Menurunkan AKI AKBRainbow DashieBelum ada peringkat
- Notulen Audit Admen (Persiapan Akreditasi)Dokumen5 halamanNotulen Audit Admen (Persiapan Akreditasi)Rainbow DashieBelum ada peringkat
- Vaginitis (Fluor Albus Non Go)Dokumen5 halamanVaginitis (Fluor Albus Non Go)Rainbow DashieBelum ada peringkat
- SOP PapsmearDokumen2 halamanSOP PapsmearRainbow DashieBelum ada peringkat
- Translate NadDokumen2 halamanTranslate NadRainbow DashieBelum ada peringkat
- Fix Translate Ke IndoDokumen3 halamanFix Translate Ke IndoRainbow DashieBelum ada peringkat
- PERKAWINANDokumen2 halamanPERKAWINANRainbow DashieBelum ada peringkat
- SK PENETAPAN INDIKATOR PRIORITAS UNTUK MONITORING FixDokumen4 halamanSK PENETAPAN INDIKATOR PRIORITAS UNTUK MONITORING FixRainbow DashieBelum ada peringkat
- Fix Translate Ke IndoDokumen3 halamanFix Translate Ke IndoRainbow DashieBelum ada peringkat
- DUPAKDokumen5 halamanDUPAKRainbow DashieBelum ada peringkat
- Soal Pu GanaDokumen8 halamanSoal Pu GanaRainbow DashieBelum ada peringkat
- Template Anung ICMEDH 2Dokumen7 halamanTemplate Anung ICMEDH 2Rainbow DashieBelum ada peringkat
- ABSTRAK Revisi Bagian AkhirDokumen1 halamanABSTRAK Revisi Bagian AkhirRainbow DashieBelum ada peringkat
- Kanker BibirDokumen4 halamanKanker BibirRainbow DashieBelum ada peringkat
- Pembatasan Energi NonDokumen6 halamanPembatasan Energi NonRainbow DashieBelum ada peringkat
- Sel ImunDokumen16 halamanSel ImunRainbow DashieBelum ada peringkat
- Temuan nEMG Pada Myotonia CongenitaDokumen10 halamanTemuan nEMG Pada Myotonia CongenitaRainbow DashieBelum ada peringkat
- Kuesioner TidurDokumen13 halamanKuesioner TidurRainbow DashieBelum ada peringkat
- Prediksi Pertumbuhan MandibulaDokumen16 halamanPrediksi Pertumbuhan MandibulaRainbow DashieBelum ada peringkat
- Pelatihan Keterampilan Motorik untuk LBP KronisDokumen5 halamanPelatihan Keterampilan Motorik untuk LBP KronisRainbow DashieBelum ada peringkat
- ERAS Pada NeonatusDokumen28 halamanERAS Pada NeonatusRainbow DashieBelum ada peringkat
- BelaNegaraSigiDokumen4 halamanBelaNegaraSigiRainbow DashieBelum ada peringkat
- Artikel B Indo Mau TerjemahanDokumen14 halamanArtikel B Indo Mau TerjemahanRainbow DashieBelum ada peringkat
- Tabel 1Dokumen3 halamanTabel 1Rainbow DashieBelum ada peringkat
- Trauma Oklusal Dikaitkan Dengan PeriodontitisDokumen9 halamanTrauma Oklusal Dikaitkan Dengan PeriodontitisRainbow DashieBelum ada peringkat
- Epidemiologi (Low Back Pain)Dokumen10 halamanEpidemiologi (Low Back Pain)Rainbow DashieBelum ada peringkat