Referat Mia
Diunggah oleh
Annastasia MiaHak Cipta
Format Tersedia
Bagikan dokumen Ini
Apakah menurut Anda dokumen ini bermanfaat?
Apakah konten ini tidak pantas?
Laporkan Dokumen IniHak Cipta:
Format Tersedia
Referat Mia
Diunggah oleh
Annastasia MiaHak Cipta:
Format Tersedia
BAB I
PENDAHULUAN
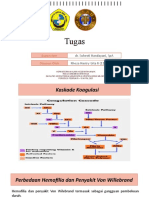
Sindroma Von Willebrand merupakan salah satu kelainan pembekuan
darah yang dapat terjadi pada anak-anak. Insidens Von Willebrand disease pada
anak dalam kepustakaan di amerika serikat dan inggris adalah 1-5 kasus baru per
100.000 anak per tahun1. Perbandingan angka kejadian pada anak laki laki dan
perempuan 1 : 2 berdasarkan fenotip, dikarenakan adanya menorrhagia dan lebih
tinggi insidens terjadinya memar1 pada anak perempuan. Faktor genetik
berperan pada patogenesis terjadinya sindroma Von Willebrand 1
Gejala atau tanda klinis utama yang sering terlihat pada anak dengan
sindroma Von Willebrand adalah mimisan atau hematoma. Perdarahan dengan
onset yang lebih lama pada luka-luka biasa, perdarahan di dalam rongga mulut,
sampai menstruasi dengan darah sangat banyak dapat ditemukan.
Gejala yang lebih berat seperti perdarahan saluran cerna jarang
ditemukan namun dapat terjadi2, pada neonatus gejala Von Willebrand disease
dapat berupa perdarahan pada saat pengguntingan tali pusat, perdarahan
konjungtiva, cephalohematoma. Dan pada anak yang sudah lebih besar dapat
berupa perdarahan pada saat sirkumsisi.
Pasien sindroma Von Willebrand biasanya datang dengan keluhan
perdarahan yang banyak dan tidak dapat berhenti. Bila perdarahan sangat
banyak dan tidak ditangani dengan segera dapat terjadi syok hingga kematian.
Anamnesis tentang riwayat keluarga tentang hal dan keluhan serupa, serta
kelainan pembekuan darah yang terjadi sebelumnya perlu ditanyakan.
Von Willebrand factor (VWF) is built up from a varying number
Annastasia Mia -Sindroma Von Willebrand 1
Kepaniteraan Klinik Ilmu Kesehatan Anak RSUD Kota Semarang
Fakultas Kedokteran Universitas Tarumanagara
of subunits, of which the larger molecules have higher haemostatic
activity. Von Willebrand disease (VWD) and thrombotic
thrombocytopenic purpura are the best known disorders with
pathognomonic changes of the highly multimerised VWF forms.
There is an established method to calculate the relative amount of
large oligomers. Our aim is to quantify the degree of VWF
multimerisation as well, to complete the densitometric analysis of
VWF electrophoresis2 (Novel evaluation method for
densitometric curves of von Willebrand factor
multimers and a new parameter (M(MW)) to
describe the degree of multimersation.
/Title
Thromb Haemost. 2009; 102(2):412-7 (ISSN: 0340-6245)
Udvardy ML; Szekeres-Csiki K; Hrsfalvi J
University of Debrecen, Debrecen, H-4012, Hungary.
Prognosis In individuals with vWD types II and III, bleeding episodes may
be severe and potentially life threatening. Individuals with type III disease who
have correspondingly low FVIII levels may develop arthropathies, as more
commonly seen in hemophilia A patients with comparably decreased FVIII levels.
The rare type 3 von Willebrand disease can manifest with severe bleeding
symptoms similar to severe hemophilia (eg, hemarthrosis, intramuscular
bleeding).
Annastasia Mia -Sindroma Von Willebrand 2
Kepaniteraan Klinik Ilmu Kesehatan Anak RSUD Kota Semarang
Fakultas Kedokteran Universitas Tarumanagara
1.Race
No influence of ethnicity on the prevalence of von Willebrand disease has been
reported.
2.Sex
von Willebrand disease affects males and females in equal numbers.
3.Age
von Willebrand disease is a congenital bleeding disorder and can be diagnosed at
any age.
BAB II
DEFINISI , EPIDEMIOLOGI , DAN ETIOLOGI
Annastasia Mia -Sindroma Von Willebrand 3
Kepaniteraan Klinik Ilmu Kesehatan Anak RSUD Kota Semarang
Fakultas Kedokteran Universitas Tarumanagara
First described by Erik Adolf von Willebrand in 1926, von Willebrand
disease is a congenital bleeding disorder characterized by a lifelong tendency
toward easy bruising, frequent epistaxis, and menorrhagia.
Von Willebrand disease (vWD) is a common, inherited, genetically and
clinically heterogeneous hemorrhagic disorder caused by a deficiency or
dysfunction of the protein termed von Willebrand factor (vWF). Consequently,
defective vWF interaction between platelets and the vessel wall impairs primary
hemostasis.2
vWF, a large, multimeric glycoprotein, circulates in blood plasma at
concentrations of approximately 10 mg/mL. In response to numerous stimuli,
vWF is released from storage granules in platelets and endothelial cells. It
performs two major roles in hemostasis. First, it mediates the adhesion of
platelets to sites of vascular injury. Second, it binds and stabilizes the
procoagulant protein factor VIII (FVIII)..3
vWD is divided into three major categories: (1) partial quantitative
deficiency (type I), (2) qualitative deficiency (type II), and (3) total deficiency
(type III). vWD type II is further divided into four variants (IIA, IIB, IIN, IIM), based
on characteristics of dysfunctional vWF. These categories correspond to distinct
molecular mechanisms, with corresponding clinical features and therapeutic
recommendations. 2
The history may reveal the following:
Increased or easy bruising
Recurrent epistaxis
Menorrhagia
Annastasia Mia -Sindroma Von Willebrand 4
Kepaniteraan Klinik Ilmu Kesehatan Anak RSUD Kota Semarang
Fakultas Kedokteran Universitas Tarumanagara
Postoperative bleeding (particularly after tonsillectomy or dental
extractions): Medical records of 99 patients younger than 18 years with
von Willebrand disease who underwent tonsillectomy were compared
with 99 patients without von Willebrand disease in the same age group;
subjects were matched for age, year of surgery, type of surgery, and
indication for surgery.[8] The study concluded that children with von
Willebrand disease have a postoperative bleeding rate similar to that of a
matched group. However, the sample size was insufficient to eliminate
any clinically important difference between the two groups.
Family history of a bleeding diathesis
Bleeding from wounds
Gingival bleeding
Postpartum bleeding
BAB III
PATOFISIOLOGI
von Willebrand disease is due to an abnormality, either quantitative or
qualitative, of the von Willebrand factor, which is a large multimeric glycoprotein
Annastasia Mia -Sindroma Von Willebrand 5
Kepaniteraan Klinik Ilmu Kesehatan Anak RSUD Kota Semarang
Fakultas Kedokteran Universitas Tarumanagara
that functions as the carrier protein for factor VIII (FVIII). [1] von Willebrand factor
is also required for normal platelet adhesion. As such, von Willebrand factor
functions in both primary (involving platelet adhesion) and secondary (involving
FVIII) hemostasis. In primary hemostasis, von Willebrand factor attaches to
platelets by its specific receptor to glycoprotein Ib on the platelet surface and
acts as an adhesive bridge between the platelets and damaged subendothelium
at the site of vascular injury. In secondary hemostasis, von Willebrand factor
protects FVIII from degradation and delivers it to the site of injury.
von Willebrand factor is composed of dimeric subunits that are linked by disulfide
bonds to form complex multimers of low, intermediate, and high molecular
weights. The small multimers function mainly as carriers for FVIII.
Highmolecular weight multimers have higher numbers of platelet-binding sites
and greater adhesive properties. Each multimeric subunit has binding sites for
the receptor glycoprotein Ib on nonactivated platelets and the receptor
glycoprotein IIb/IIIa on activated platelets. This facilitates both platelet adhesion
and platelet aggregation, making high molecular weight multimers most
important for normal platelet function.
Annastasia Mia -Sindroma Von Willebrand 6
Kepaniteraan Klinik Ilmu Kesehatan Anak RSUD Kota Semarang
Fakultas Kedokteran Universitas Tarumanagara
von Willebrand disease types
von Willebrand disease can be classified into 3 main types.
Type 1 von Willebrand disease, which accounts for 70-80% of cases, is
characterized by a partial quantitative decrease of qualitatively normal
von Willebrand factor and FVIII. An individual with type 1 von Willebrand
disease generally has mild clinical symptoms, and this type is usually
inherited as an autosomal dominant trait; however, penetrance may
widely vary in a single family. In addition, clinical and laboratory findings
may vary in the same patient on different occasions. Typically, a
proportional reduction in von Willebrand factor activity, von Willebrand
factor antigen, and FVIII is observed in type 1 von Willebrand disease. [2]
Annastasia Mia -Sindroma Von Willebrand 7
Kepaniteraan Klinik Ilmu Kesehatan Anak RSUD Kota Semarang
Fakultas Kedokteran Universitas Tarumanagara
Type 2 disease accounts for 15-20% of von Willebrand disease cases. Type
2 von Willebrand disease is a variant of the disease with primarily
qualitative defects of von Willebrand factor. Type 2 von Willebrand
[3]
disease can be either autosomal dominant or autosomal recessive. Of
the 4 described type 2 von Willebrand disease subtypes (ie, 2A, 2B, 2M,
2N), type 2A von Willebrand disease is by far the most common.
Type 2A von Willebrand disease is inherited as an autosomal
dominant trait and is characterized by normal-to-reduced plasma
levels of factor VIIIc (FVIIIc) and von Willebrand factor. Analysis of
von Willebrand factor multimers reveals a relative reduction in
intermediate and high molecular weight multimer complexes. The
multimeric abnormalities are commonly the result of in vivo
proteolytic degradation of the von Willebrand factor. The
ristocetin cofactor activity is greatly reduced, and the platelet von
Willebrand factor reveals multimeric abnormalities similar to
those found in plasma.
Type 2B von Willebrand disease is also an autosomal dominant
trait. This type is characterized by a reduction in the proportion of
high molecular weight von Willebrand factor multimers, whereas
the proportion of lowmolecular weight fragments are increased.
Patients with type 2B von Willebrand disease have a hemostatic
defect caused by a qualitatively abnormal von Willebrand factor
and intermittent thrombocytopenia. This is a gain of function
mutation in the von Willebrand factor causing spontaneous
binding to platelets and rapid clearance of both platelets and high
molecular weight von Willebrand factor multimers. The abnormal
von Willebrand factor has an increased affinity for platelet
glycoprotein Ib.
Annastasia Mia -Sindroma Von Willebrand 8
Kepaniteraan Klinik Ilmu Kesehatan Anak RSUD Kota Semarang
Fakultas Kedokteran Universitas Tarumanagara
The platelet count may fall further during pregnancy, in
association with surgical procedures, or after the administration of
desmopressin acetate (DDAVP). Although some investigators
found DDAVP to be clinically useful in persons with type 2B von
Willebrand disease , studies directed at excluding the 2B variant
should be completed before DDAVP is used. Measurements of
FVIIIc and von Willebrand factor in plasma vary; however, studies
involving the use of titered doses of ristocetin reveal that
aggregation of normal platelets is enhanced and induced by
unusually small amounts of the drug (low dose ristocetin induced
platelet aggregation assay- LD- RIPA).
In patients with the rare type 2M von Willebrand disease,
laboratory results are similar to those of certain patients with type
2A von Willebrand disease. Type 2M von Willebrand disease is
characterized by a decreased platelet-directed function that is not
due to a decrease of highmolecular weight multimers. Laboratory
findings show decreased von Willebrand factor activity, but von
Willebrand factor antigen, FVIII, and multimer analysis are found
to be within reference range. This is caused by a defect in the von
Willebrand factor gene that produces decreased or absent binding
to platelet glycoprotein1b and is autosomal in inheritance.
Type 2N von Willebrand disease is also rare and is characterized by
a markedly decreased affinity of von Willebrand factor for FVIII
because of a genetic mutation in the factor VIII binding region of
von Willebrand factor, resulting in FVIII levels reduced to usually
around 5% of the reference range. Other von Willebrand factor
laboratory parameters (ie, von Willebrand factor antigen
[VWF:Ag], ristocetin cofactor activity) are usually normal. The
Annastasia Mia -Sindroma Von Willebrand 9
Kepaniteraan Klinik Ilmu Kesehatan Anak RSUD Kota Semarang
Fakultas Kedokteran Universitas Tarumanagara
FVIII-binding defect in these patients is inherited in an autosomal
recessive manner. Evaluate patients with FVIII deficiency (mild
FVIII deficiency) and a bleeding disorder that is not clearly
transmitted as an X-linked disorder or those who respond
incompletely to hemophilia A therapy for type 2N von Willebrand
disease. Unfortunately, the confirmatory test which is a specific
assay of FVIII binding to von Willebrand factor for type 2N von
Willebrand disease is not routinely available, likely resulting in an
underestimate of the true frequency of this subtype.[4]
Type 3 is the most severe form of von Willebrand disease. In the homozygous
patient, type 3 von Willebrand disease is characterized by marked deficiencies of
both von Willebrand factor and FVIIIc in the plasma, the absence of von
Willebrand factor from both platelets and endothelial cells, and a lack of
response to DDAVP. Type 3 von Willebrand disease is characterized by severe
clinical bleeding and is inherited as an autosomal recessive trait. Consanguinity is
common in kindreds with this variant. Less severe clinical abnormalities and
laboratory abnormalities may be identified in occasional heterozygotes; however,
such cases are difficult to identify. Multimeric analysis of the small amount of von
Willebrand factor present yields variable results, in some cases revealing only
small multimers.
BAB IV
MANIFESTASI KLINIS
Annastasia Mia -Sindroma Von Willebrand 10
Kepaniteraan Klinik Ilmu Kesehatan Anak RSUD Kota Semarang
Fakultas Kedokteran Universitas Tarumanagara
Many children with von Willebrand disease (VWD) are asymptomatic and are
diagnosed as a result of a positive family history or during routine preoperative
screening . Some children may have completely normal screening and bleed at
procedures in areas with increased fibrinolysis such as the mouth i.e.
tonsillectomy. Importantly, remember that a wide variation in clinical
manifestations is observed, even for members of the same family.
The diagnosis of von Willebrand disease can be challenging and depends on an
accurate personal and family bleeding history, as well as demonstration of a low
von Willebrand factor (VWF) level.[6, 7]
The history may reveal the following:
Increased or easy bruising
Recurrent epistaxis
Menorrhagia
Postoperative bleeding (particularly after tonsillectomy or dental
extractions): Medical records of 99 patients younger than 18 years with
von Willebrand disease who underwent tonsillectomy were compared
with 99 patients without von Willebrand disease in the same age group;
subjects were matched for age, year of surgery, type of surgery, and
indication for surgery.[8] The study concluded that children with von
Willebrand disease have a postoperative bleeding rate similar to that of a
matched group. However, the sample size was insufficient to eliminate
any clinically important difference between the two groups.
Family history of a bleeding diathesis
Bleeding from wounds
Gingival bleeding
Postpartum bleeding
Annastasia Mia -Sindroma Von Willebrand 11
Kepaniteraan Klinik Ilmu Kesehatan Anak RSUD Kota Semarang
Fakultas Kedokteran Universitas Tarumanagara
BAB V
PEMERIKSAAN FISIK DAN PEMERIKSAAN PENUNJANG
Screening tests for von Willebrand disease (VWD) include the following:
1. Laboraturium:
CBC count: Assess platelet number and morphology, which should be
normal in most patients with von Willebrand disease, except those with
type 2B von Willebrand disease who may have thrombocytopenia.
Template bleeding time: Because it is reasonably well standardized, the
template bleeding time is used as a screening test for primary hemostasis.
The reference range for the bleeding time in children is longer than that
of adults. Results of the bleeding time are affected by many technical
factors, such as the direction of the incision and the skill of the technician.
Although a bleeding time outside of the reference range may suggest a
defect in hemostasis, it is not diagnostic. Similarly, a bleeding time within
the reference range does not exclude the presence of such a defect.
Although neither sensitive nor specific for von Willebrand disease,
template-bleeding time is outside of the reference range in about 50% of
patients with type 1 von Willebrand disease. Patients with von Willebrand
disease types 2A, 2B, 2M, and 3 often have prolonged bleeding times. The
template bleeding time has largely been replaced by automatic platelet
function analyzers (PFAs) such as the PFA-100.
Prothrombin time (PT) is within reference range in von Willebrand
disease.
Activated partial thromboplastin time (aPTT): Approximately 25% of
patients with type 1 von Willebrand disease have aPTT results outside of
Annastasia Mia -Sindroma Von Willebrand 12
Kepaniteraan Klinik Ilmu Kesehatan Anak RSUD Kota Semarang
Fakultas Kedokteran Universitas Tarumanagara
the reference range. These results may be caused by concurrent
deficiencies of other clotting factors in addition to, or rather than, factor
VIII (FVIII). The aPTT should be outside of the reference range in patients
with severe von Willebrand disease or type 2N von Willebrand disease in
whom circulating FVIII levels are very low. Because aPTT and the template
bleeding time are insensitive tests for von Willebrand disease, add von
Willebrand factor (VWF) activity to the screening tests performed for
patients with suspected bleeding disorders (see below).
Specific assays include the following:
von Willebrand factor levels: von Willebrand factor levels vary and can be
influenced by numerous factors including blood type. Individuals with
type O blood have lower values of von Willebrand factor levels on
average, whereas those with type AB blood have higher values of von
Willebrand factor. Day-to-day variation in von Willebrand factor levels is a
normal occurrence in the same individual; therefore, a single level within
reference range does not exclude the diagnosis of von Willebrand disease.
Also, estrogen levels increase von Willebrand factor and may affect results
in adolescent females and women with menorrhagia.
FVIII activity: FVIII activity is variably decreased.
von Willebrand factor activity (ristocetin cofactor): Ristocetin is an
antibiotic that causes von Willebrand factor to bind to and, subsequently,
to activate platelets. In the ristocetin cofactor assay, platelets from
individuals who are healthy, standard concentrations of ristocetin, and
varying quantities of patient or control plasma are used. In individuals
who are healthy, platelets rapidly agglutinate in response to ristocetin;
however, the presence of plasma von Willebrand factor is necessary for
the reaction to occur. The degree of platelet agglutination is proportional
Annastasia Mia -Sindroma Von Willebrand 13
Kepaniteraan Klinik Ilmu Kesehatan Anak RSUD Kota Semarang
Fakultas Kedokteran Universitas Tarumanagara
to the concentration of von Willebrand factor in the plasma. Several
variations of this assay have been developed. Because the result of this
assay reflects the functional activity of von Willebrand factor, it is usually
called the von Willebrand factor activity. It is variably decreased in von
Willebrand disease.
von Willebrand factor antigen: The total plasma concentration of von
Willebrand factor protein is measured by one of several assays. The
Laurell rocket immunoelectrophoresis technique measures the amount of
von Willebrand factor protein in the plasma, whereas radioimmunoassays
and enzyme-linked immunoabsorbent assays reflect the number of von
Willebrand factorbinding sites. These tests determine the total amount
of von Willebrand factor antigen in the plasma but do not reflect its
molecular structure and, hence, may be normal in von Willebrand disease
variants with abnormal multimers. Therefore, von Willebrand factor
antigen is variably decreased.
In multimer analysis to determine the physical structure of von Willebrand factor
(ie, whether high molecular weight multimers are present), plasma is
electrophoresed through agarose gel. The presence or absence of high molecular
weight von Willebrand factor is used to classify von Willebrand disease. Absence
or decreased levels of high molecular weight von Willebrand factor multimers is
consistent with type 2 von Willebrand disease. Further analysis of von Willebrand
factor subunits has been performed with sophisticated electrophoretic
techniques, resulting in the description of many type 2 variants.
In some laboratories, platelet von Willebrand factor analysis is performed. Gene
analysis can also be performed for diagnosis.
Annastasia Mia -Sindroma Von Willebrand 14
Kepaniteraan Klinik Ilmu Kesehatan Anak RSUD Kota Semarang
Fakultas Kedokteran Universitas Tarumanagara
BAB VI
DIAGNOSIS
Diagnosis ditegakkan berdasarkan anamnesis, pemeriksaan fisik dan
pemeriksaan penunjang.3
Anamnesis
Keluhan yang sering ditemukan adalah bengkak di kedua kelopak
mata, perut, tungkai, atau seluruh tubuh, dan dapat disertai jumlah urin
yang berkurang. Keluhan lain juga dapat ditemukan seperti urin berwarna
kemerahan.3
Annastasia Mia -Sindroma Von Willebrand 15
Kepaniteraan Klinik Ilmu Kesehatan Anak RSUD Kota Semarang
Fakultas Kedokteran Universitas Tarumanagara
Pemeriksaan fisis
Pada pemeriksaan fisik sindrom nefrotik dapat ditemukan edema
di kedua kelopak mata, tungkai, atau adanya asites dan edema
skrotum/labia. Kadang-kadang ditemukan hipertensi3
Pemeriksaan penunjang
Pada urinalisis ditemukan proteinuria masif (3+ sampai 4+) (> 40 mg/m2
LPB/jam atau 50 mg/kg/hari atau rasio protein/kreatinin pada urin sewaktu >
2 mg/mg atau dipstik 2+), dapat disertai hematuria. Pada pemeriksaan
darah didapatkan hipoalbuminemia (< 2,5 g/dl), hiperkolesterolemia(> 200
mg/dL), dan laju endap darah yang meningkat, rasio albumin/globulin
terbalik. Kadar ureum dan kreatinin umumnya normal kecuali ada penurunan
fungsi ginjal. Bila terjadi hematuria mikroskopik (>20 eritrosit/LPB) dicurigai
adanya lesi glomerular (mis. Sclerosis glomerulus fokal). 3
Batasan :1
1. Remisi : Proteinuria (-) (proteinuria < 4mg / m 2LPB / jam)
.............................3 hari berturut turut dalam 1 minggu
2. Relaps : Proteinuria >/ 2+ ( proteinuria > 40 mg / m 2 LPB /
.............................jam) 3 hari.berturut turut dalam 1 minggu.
3. Relaps jarang : Relaps kurang dari 2x dalam 6 bulan pertama
.............................setelah respons awal atau kurang dari 4x per tahun
.............................pengamatan.
4. Relaps sering : Relaps >/ 2x dalam 6 bulan pertama setelah
..............................responsawal atau >/4x dalam periode 1 tahun
Annastasia Mia -Sindroma Von Willebrand 16
Kepaniteraan Klinik Ilmu Kesehatan Anak RSUD Kota Semarang
Fakultas Kedokteran Universitas Tarumanagara
5. Dependen steroid : Relaps 2x berurutan pada saat dosis steroid
................................ diturunkan (alternating) atau dalam 14 hari
setelah ..............................pengobatan dihentikan.
6. Resisten steroid : Tidak terjadi remisi pada pengobatan prednison
.................................dosis penuh ( full dose ) 2mg/ kgbb/ hari selama
4 ..............................minggu.
7. Sensitif steroid : Remisi terjadi pada pemberian prednison dosis
.............................penuh selama 4 minggu.
BAB VII
PENATALAKSANAAN
Tatalaksana Umum1
Evidence-based guidelines for the diagnosis and management of von
Willebrand disease (VWD) have been established.[10, 11]
Minor bleeding problems in patients with von Willebrand disease, such as
bruising or a brief nosebleed, may not require specific treatment. For more
serious bleeding, medications that can raise the von Willebrand factor (VWF)
level and, thereby, limit bleeding are available. The goal of therapy is to correct
the defect in platelet adhesiveness (by raising the level of effective von
Annastasia Mia -Sindroma Von Willebrand 17
Kepaniteraan Klinik Ilmu Kesehatan Anak RSUD Kota Semarang
Fakultas Kedokteran Universitas Tarumanagara
Willebrand factor) and the defect in blood coagulation (by raising the factor VIII
[FVIII] level). In recent years, desmopressin (1-deamine-8-D-arginine
vasopressin [DDAVP]) has become a mainstay of therapy for most patients with
mild von Willebrand disease. At appropriate doses, DDAVP causes a 2-fold to 5-
fold increase in plasma von Willebrand factor and FVIII concentrations in
individuals who are healthy and patients who are responsive. DDAVP can be
used to treat bleeding complications or to prepare patients with von
Willebrand disease for surgery.
In general, a patient's responsiveness to DDAVP prior to its use for these
purposes can be determined. Once determined, such responsiveness is
generally consistent in patients over time and within families. In patients with
serious bleeding, prompt treatment is important in order to decrease the
possibility of complications.
Remember that in type IIB von Willebrand disease, DDAVP may cause a
paradoxical drop in the platelet count and should not be used in a therapeutic
setting without prior testing to see how the patient responds.
Vasopressin analogues
Class Summary
Desmopressin is a synthetic analogue of antidiuretic hormone. It is considered
the primary treatment for bleeding in individuals with mild von Willebrand
disease (VWD). It works by causing release of von Willebrand factor (VWF) from
endothelial storage sites.
Desmopressin can be administered intravenously, intranasally, or
subcutaneously. The dose for hemostasis is approximately 15 times the dosage
used to treat individuals with diabetes insipidus. The regular intranasal
preparation (0.1 mg/mL), which is used to treat persons with diabetes
Annastasia Mia -Sindroma Von Willebrand 18
Kepaniteraan Klinik Ilmu Kesehatan Anak RSUD Kota Semarang
Fakultas Kedokteran Universitas Tarumanagara
insipidus, is too dilute to elicit a hemostatic response. A high-concentration
intranasal preparation (ie, Stimate 1.5 mg/mL) has been licensed and has
shown a similar response as the intravenous form.
The higher concentration intranasal preparation allows home treatment for
bleeding symptoms; however, experience with its use in the surgical setting is
limited. Most experience in treating individuals with von Willebrand disease is
with intravenous infusion, with which the response is rapid (ie, peak von
Willebrand factor levels in approximately 45-90 min of infusion). Doses may be
repeated at intervals of 12-24 hours for continued bleeding or for postoperative
use. Desmopressin has also been administered subcutaneously with a favorable
response.
View full drug information
Desmopressin (Stimate)
Increases cellular permeability of collecting ducts, resulting in reabsorption of
water by kidneys.
Test patients for response prior to usage in a bleeding episode. A 2-fold to 5-
fold increase in VWF and FVIII commonly is obtained after treatment.
The higher concentration of desmopressin (ie, Stimate 1.5 mg/mL) is prescribed
for VWD to provide an adequate dose.
Plasma products
Class Summary
For patients with von Willebrand disease who do not respond to desmopressin,
and for individuals with the rare types 2B or 3 von Willebrand disease, plasma-
derived factor VIII (FVIII) concentrates that contain von Willebrand factor in
Annastasia Mia -Sindroma Von Willebrand 19
Kepaniteraan Klinik Ilmu Kesehatan Anak RSUD Kota Semarang
Fakultas Kedokteran Universitas Tarumanagara
high molecular weight can be used. The product used must contain von
Willebrand factor in the highmolecular weight form to be effective. However,
most available FVIII concentrates do not contain sufficient von Willebrand
factor to be used in von Willebrand disease. Cryoprecipitate contains
multimeric von Willebrand factor; however, concerns about possible virus
transmission have led many clinicians to choose FVIII products that contain
multimeric von Willebrand factor and have undergone viral inactivation
processes.
Only a minority of currently available FVIII products contain von Willebrand
factor; the protein has been eliminated from the others. In general, the dosage
of cryoprecipitate or FVIII to be used is calculated on the basis of FVIII units.
Other blood products are rarely required for patients with von Willebrand
disease. Platelet transfusion may benefit patients with type 3 von Willebrand
disease or platelet-type von Willebrand disease who do not respond to von
Willebrand factorcontaining concentrates or cryoprecipitate.
Antihemophilic factor/von Willebrand Factor Complex, human (Alphanate,
Humate-P, Wilate)
Some FVIII concentrates (eg, Humate-P, Alphanate, Wilate) also contain VWF in
high molecular weight form. These concentrates are especially useful in types
2B and 3 vWD.
Alphanate is indicated to prevent excessive bleeding for surgical and invasive
procedures in vWD in cases in which desmopressin is either ineffective or
contraindicated. It is not indicated for patients with severe vWD (ie, Type 3)
undergoing major surgery.
Annastasia Mia -Sindroma Von Willebrand 20
Kepaniteraan Klinik Ilmu Kesehatan Anak RSUD Kota Semarang
Fakultas Kedokteran Universitas Tarumanagara
Humate-P is indicated for treatment and prevention of spontaneous and
trauma-induced bleeding episodes for patients with mild-to-moderate or
severe vWD.
View full drug information
Aminocaproic acid (Amicar)
Inhibits fibrinolysis via inhibition of plasminogen activator substances and, to a
lesser degree, through antiplasmin activity. Main problem is that the thrombi
that forms during treatment are not lysed and effectiveness is uncertain. Has
been used to prevent recurrence of subarachnoid hemorrhage (SAH). Useful in
mucous membrane bleeding.
BAB VII
PROGNOSIS
ndividuals with von Willebrand disease have a lifelong tendency toward
easy bruising, frequent epistaxis, and menorrhagia.
BAB IX
PENUTUP
Annastasia Mia -Sindroma Von Willebrand 21
Kepaniteraan Klinik Ilmu Kesehatan Anak RSUD Kota Semarang
Fakultas Kedokteran Universitas Tarumanagara
Telah dibicarakan penyakit sindroma nefrotik yang merupakan penyakit
ginjal glomerolus yang terbanyak, khususnya pada anak. Sindrom nefrotik (SN)
adalah keadaan klinis yang ditandai dengan proteinuria masif (terutama albumin)
(>40 mg/m2/jam); hipoproteinemia (albumin serum <3,0 g/dL);
hiperkolesterolemia ( >250 mg/dL); dan edema. Kelainan mendasar pada
sindroma nefrotik adalah peningkatan permeabilitas dinding kapiler glomerulus
yang mengakibatkan proteinuria masif dan hipoalbuminemia.
Karena lebih dari 80% anak berusia di bawah 13 tahun termasuk SN
sensitif steroid (terutama SNKM), maka terapi steroid dapat dimulai tanpa
didahului biopsi ginjal bila anak menunjukkan gambaran klinis yang sesuai
dengan SN. Lebih dari 90% memperlihtkan respons yang baik dalam 4 minggu.
Pasien SNKM yang resisten steroid atau mengalami relaps memerlukan tambahan
terapi imunosupresif lain. Terapi agresif untuk SN kongenital dengan nefrektomi
dini, dialisis, dan transplantasi adalah satu-satunya terapi yang efektif.
Prognosa pada yang yang berespon baik terhadap pengobatan steroid
sangat jarang berkembang menjadi gagal ginjal terminal . Tetapi pada pasien
yang tidak berespon dengan steroid / resisten steroid dapat berkembang menjadi
gagal ginjal terminal yang membutuhkan dialisis dan transplantasi ginjal.
DAFTAR PUSTAKA
1. Konsensus Ikatan Dokter Anak Indonesia. Tatalaksana sindrom nefrotik
idiopatik pada anak. Edisi ke-2 Cetakan kedua Jakarta : Badan Penerbit
Ikatan Dokter Anak Indonesia; 2012.h.1-20.
Annastasia Mia -Sindroma Von Willebrand 22
Kepaniteraan Klinik Ilmu Kesehatan Anak RSUD Kota Semarang
Fakultas Kedokteran Universitas Tarumanagara
2. Pais Priya, Avner ED. Nephrotic syndrome. Dalam: Kliegman RM, Behrman
RE, Jenson HB, Stanton BF, penyunting. Nelson textbook of pediatrics.
Edisi ke-19.Philadelphia: Saunders;2011.h.1801-7.
3. Husein A, dkk. Buku Ajar Nefrologi Anak. Edisi kedua. Jakarta : Ikatan
Dokter Anak Indonesia; 2010. hal 381-421
4. Pudjiadi AH, dkk. Pedoman Pelayanan Medis . Jilid satu. Jakarta: Ikatan
Dokter Anak Indonesia; 2010. Hal 274-6
5. Suyitno H,dkk. Pedoman Imunisasi Di Indonesia. Edisi empat. Jakarta:
Ikatan Dokter Anak Indonesia;2011. Hal 307-16
Annastasia Mia -Sindroma Von Willebrand 23
Kepaniteraan Klinik Ilmu Kesehatan Anak RSUD Kota Semarang
Fakultas Kedokteran Universitas Tarumanagara
Anda mungkin juga menyukai
- Mikrobiologi Perubatan I: Patogen dan Mikrobiologi ManusiaDari EverandMikrobiologi Perubatan I: Patogen dan Mikrobiologi ManusiaPenilaian: 2.5 dari 5 bintang2.5/5 (2)
- Mikrobiologi Medis I: Patogen dan Mikrobioma ManusiaDari EverandMikrobiologi Medis I: Patogen dan Mikrobioma ManusiaPenilaian: 4 dari 5 bintang4/5 (11)
- Von WillebrandDokumen18 halamanVon WillebrandRizcky Naldy Eka Putra100% (1)
- Von WillebrandDokumen18 halamanVon WillebrandRizcky Naldy Eka PutraBelum ada peringkat
- Von Willebrand DiseaseDokumen9 halamanVon Willebrand DiseaseAnonymous qDteDz0% (1)
- Von Willebrand DiseaseDokumen3 halamanVon Willebrand DiseaseMuhammad Mardhiya AlghifariBelum ada peringkat
- Kelainan Darah Pada AnjingDokumen5 halamanKelainan Darah Pada Anjingasyrafun nisaBelum ada peringkat
- Penyakit Von WillebrandDokumen4 halamanPenyakit Von WillebrandOMFS Unair 19Belum ada peringkat
- Von Willebrand Disease: ProkoagulanDokumen7 halamanVon Willebrand Disease: ProkoagulanSetywanty LayuklinggiBelum ada peringkat
- VWD BenarDokumen19 halamanVWD BenarAjeng DwintaBelum ada peringkat
- Penyakit Von WillebrandDokumen2 halamanPenyakit Von WillebrandChiki CacaBelum ada peringkat
- Von Willebrand DiseaseDokumen11 halamanVon Willebrand Diseaseaudina fajrianaBelum ada peringkat
- Penyakit Von WillebrandDokumen2 halamanPenyakit Von WillebrandEvans Oktora RolindriBelum ada peringkat
- Penyakit Von WillebrandDokumen9 halamanPenyakit Von WillebrandaaaaishBelum ada peringkat
- Hemofilia Dan Von Willebrand Disease: Dr. Nice Rachmawati Masnadi, Spa (K)Dokumen18 halamanHemofilia Dan Von Willebrand Disease: Dr. Nice Rachmawati Masnadi, Spa (K)sonnya morisaBelum ada peringkat
- 028 - Nathin Loria Saragih - ADokumen2 halaman028 - Nathin Loria Saragih - AJonathanBelum ada peringkat
- IDK Penyakit Von WillebrandDokumen3 halamanIDK Penyakit Von WillebrandAnna AndanyBelum ada peringkat
- Refrat PVWDokumen8 halamanRefrat PVWJupiter CarlBelum ada peringkat
- Definisi Dan Patofisiologi VWDDokumen6 halamanDefinisi Dan Patofisiologi VWDBadzlinaKhairunizzahraBelum ada peringkat
- Penyakit Von WillebrandDokumen5 halamanPenyakit Von WillebrandSepta Ike GayatriBelum ada peringkat
- Von WillebrandDokumen15 halamanVon WillebrandTerry ReedBelum ada peringkat
- Penyakit Von WillebrandDokumen7 halamanPenyakit Von Willebrandafinarosida05Belum ada peringkat
- Von Willebrand Disease: Revky Akbar Iztian 1810211102Dokumen13 halamanVon Willebrand Disease: Revky Akbar Iztian 1810211102Makhluk BarBarBelum ada peringkat
- Bahan Tutorial Minggu 4 2.1 (VWD, Hemofilia, VKDB)Dokumen6 halamanBahan Tutorial Minggu 4 2.1 (VWD, Hemofilia, VKDB)Putriku fatiyaBelum ada peringkat
- Penyakit Von WillebrandDokumen4 halamanPenyakit Von WillebrandOcta TirandhaBelum ada peringkat
- Makalah Penyakit Von WillebrandDokumen11 halamanMakalah Penyakit Von Willebrandindah ayu50% (2)
- Penyakit Von WillebrandDokumen1 halamanPenyakit Von WillebrandAdinda RaBelum ada peringkat
- Penyakit Von WilbrandDokumen15 halamanPenyakit Von Wilbrandronianandaperwira_haBelum ada peringkat
- LTM 2 Penyakit Von Willebrand Dan Defisiensi Vitamin KDokumen4 halamanLTM 2 Penyakit Von Willebrand Dan Defisiensi Vitamin KRaymondRumantirWardhanaBelum ada peringkat
- Von Willebrand Dan HemofiliaDokumen6 halamanVon Willebrand Dan HemofiliaRheza GitaaBelum ada peringkat
- Diathesis Hemoragik - DK2Dokumen7 halamanDiathesis Hemoragik - DK2Nabilla Puteri TrisiraBelum ada peringkat
- Penyakit Von WillebrandDokumen23 halamanPenyakit Von WillebrandImanuelBelum ada peringkat
- Von Willebrand DiseaseDokumen18 halamanVon Willebrand DiseaserezaatqiaBelum ada peringkat
- Sindrom Wiskott AldrichDokumen7 halamanSindrom Wiskott AldrichImut MainahBelum ada peringkat
- Kelainan VaskulerDokumen2 halamanKelainan VaskulerKandela FazriatrisuciBelum ada peringkat
- Mahesa TarunaDokumen25 halamanMahesa TarunaimamunnasBelum ada peringkat
- Nefroblastoma (Tumor Wilms)Dokumen30 halamanNefroblastoma (Tumor Wilms)Elaina Ross0% (1)
- Makalah Kasus Coagulation DisorderDokumen25 halamanMakalah Kasus Coagulation DisorderAfif PusamaniaBelum ada peringkat
- Askep VaricellaDokumen14 halamanAskep VaricellaLailaAnggraini50% (2)
- Sindrom WiskotaldrhDokumen7 halamanSindrom WiskotaldrhMuhammad AkrimBelum ada peringkat
- Veruka Vulgaris (Silvani)Dokumen11 halamanVeruka Vulgaris (Silvani)Silvani HamsyahBelum ada peringkat
- LP Pembekuan DarahDokumen11 halamanLP Pembekuan Darahlurfi minilBelum ada peringkat
- LP & Askep AMLDokumen33 halamanLP & Askep AMLancours100% (3)
- Askep VariselaDokumen13 halamanAskep VariselaRAHMA AYU MULIAWATI 011Belum ada peringkat
- Vaksin Varicella ZosterDokumen11 halamanVaksin Varicella ZosterTuti DamBelum ada peringkat
- Hemofilia & VonwillebrandDokumen46 halamanHemofilia & VonwillebrandkynatroemanBelum ada peringkat
- Platelet DisorderDokumen45 halamanPlatelet DisorderHarlisa Puspa WatiBelum ada peringkat
- HemofiliaDokumen13 halamanHemofiliaLudi NugrohoBelum ada peringkat
- ThrombopheblitisDokumen2 halamanThrombopheblitisALIFIA ADILA ASMARABelum ada peringkat
- Askep All Ni Putu Wisma DewiDokumen58 halamanAskep All Ni Putu Wisma DewiI G. Ayu Putu Satya LaksmiBelum ada peringkat
- ItpDokumen18 halamanItpEurasia BlossomBelum ada peringkat
- Varisella FixDokumen27 halamanVarisella Fixanggi purnamasariBelum ada peringkat
- LP Varicella 072759Dokumen29 halamanLP Varicella 072759elisa sabetBelum ada peringkat
- Von Willebrand DiseaseDokumen17 halamanVon Willebrand Diseaseayutiarapratiwi100% (1)
- LP AmlDokumen13 halamanLP AmlNindy YuliawatiBelum ada peringkat
- Tugas Leukosit Dan Kanker UsusDokumen20 halamanTugas Leukosit Dan Kanker UsusMuhammad Ali Faqi SarifuddinBelum ada peringkat
- Makalah PBL D4 - Sken 8Dokumen10 halamanMakalah PBL D4 - Sken 8Michael LeanielBelum ada peringkat
- Coronavirus Covid-19. Membela diri. Cara menghindari penularan. Bagaimana melindungi keluarga dan pekerjaan Anda. Diperbarui edisi keempat.Dari EverandCoronavirus Covid-19. Membela diri. Cara menghindari penularan. Bagaimana melindungi keluarga dan pekerjaan Anda. Diperbarui edisi keempat.Penilaian: 5 dari 5 bintang5/5 (2)