Anda mungkin juga menyukai
- Pabk UpdateDokumen22 halamanPabk Updatediary mahasiswa100% (1)
- Gangguan Mineral Dan Tulang Pada Gagal Ginjal KronisDokumen16 halamanGangguan Mineral Dan Tulang Pada Gagal Ginjal Kronisjason wotavizkanBelum ada peringkat
- Perawatan Pada Kelompok Rentan Saat BencanaDokumen25 halamanPerawatan Pada Kelompok Rentan Saat Bencanadewa80% (30)
- MENINGOCELEDokumen29 halamanMENINGOCELEyesi_widyastutiBelum ada peringkat
- LP CMLDokumen18 halamanLP CMLdewiretnoBelum ada peringkat
- LP Hipofise Siap KirimDokumen40 halamanLP Hipofise Siap Kirimvenny riska wulan cahyaniBelum ada peringkat
- Cerebral Sinus Venosus ThrombosisDokumen10 halamanCerebral Sinus Venosus ThrombosisTejo Pramono100% (1)
- Retinoblastoma Pada AnakDokumen23 halamanRetinoblastoma Pada AnakMohamad FaisalBelum ada peringkat
- Makalah MiokarditisDokumen15 halamanMakalah MiokarditisNurmala MalaBelum ada peringkat
- Enny Patofisiologi Syok SeptikDokumen1 halamanEnny Patofisiologi Syok SeptikEnny Suryanti0% (1)
- Konsep Dasar Penyakit TalasemiaDokumen23 halamanKonsep Dasar Penyakit TalasemiariskaBelum ada peringkat
- Tutorial HemofiliaDokumen19 halamanTutorial HemofiliaWayanBelum ada peringkat
- LP DMDokumen14 halamanLP DMCahyo AdhiBelum ada peringkat
- Limfoma BurkittDokumen2 halamanLimfoma BurkittDinda yuliasariBelum ada peringkat
- LP Anemia HemolitikDokumen9 halamanLP Anemia HemolitikJayanti IndrayaniBelum ada peringkat
- Bab II Crs Paralisis HipokalemiDokumen25 halamanBab II Crs Paralisis HipokalemiAyuindiraBelum ada peringkat
- LP TidurDokumen14 halamanLP TiduriyanBelum ada peringkat
- Systemic Lupus ErythematosusDokumen12 halamanSystemic Lupus ErythematosusPutri KartikaBelum ada peringkat
- LP Cva IchDokumen23 halamanLP Cva IchPutri UtomoBelum ada peringkat
- Uveal Tract Adalah Komponen Vascular Utama Pada MataDokumen9 halamanUveal Tract Adalah Komponen Vascular Utama Pada MataZurya UdayanaBelum ada peringkat
- Laporan Pendahuluan Idiopatik Trombositopenia PurpuraDokumen13 halamanLaporan Pendahuluan Idiopatik Trombositopenia PurpuraRizka Ayu HikmawahBelum ada peringkat
- Aging ProcessDokumen9 halamanAging ProcessRandi N. FarizaBelum ada peringkat
- Askep Thalasemia Pada AnakDokumen95 halamanAskep Thalasemia Pada AnakYohana DestiaBelum ada peringkat
- Makalah Gangguan Sistem Hematologi Askep Anemia Defisiensi BesiDokumen23 halamanMakalah Gangguan Sistem Hematologi Askep Anemia Defisiensi BesiPhiedta Purnamasari RahmadhaniBelum ada peringkat
- Polisitemia VeraDokumen27 halamanPolisitemia VeraMuhammad AgrifianBelum ada peringkat
- Jurnal ThalasemiaDokumen15 halamanJurnal ThalasemiaSulpianah Matchjhoo Adinul IslamBelum ada peringkat
- LP EncephalitisDokumen13 halamanLP Encephalitisjaya saputraBelum ada peringkat
- Anatomi Fisiologi PlasentaDokumen6 halamanAnatomi Fisiologi PlasentaHildaBelum ada peringkat
- Patogenesis ThalasemiaDokumen2 halamanPatogenesis Thalasemiafile file100% (1)
- Jejas Sel Dan AdaptasiDokumen60 halamanJejas Sel Dan AdaptasilolaaputriiBelum ada peringkat
- Tugas KMB 1 AnemiaDokumen38 halamanTugas KMB 1 AnemiaRimesaBelum ada peringkat
- KEPUTIHANDokumen55 halamanKEPUTIHANMuzayyinatul HayatBelum ada peringkat
- Patofisiologi Sindrom DownDokumen2 halamanPatofisiologi Sindrom Downbella0% (1)
- Penilaian KramerDokumen3 halamanPenilaian KramerRizah Istighfari FauziaBelum ada peringkat
- Ca TiroidDokumen21 halamanCa TiroidHemalatha PathmanathanBelum ada peringkat
- Makalah Imun Biologi TransfusiDokumen34 halamanMakalah Imun Biologi TransfusiMelvin PhilipsBelum ada peringkat
- Terapi Aktivitas KelompokDokumen17 halamanTerapi Aktivitas Kelompoksoffi whyuniBelum ada peringkat
- EMFISEMADokumen15 halamanEMFISEMANuryana Siti MariyamBelum ada peringkat
- Patofisiologi Stroke EmboliDokumen2 halamanPatofisiologi Stroke EmboliFerly AnasBelum ada peringkat
- Pemfis ThalasemiaDokumen2 halamanPemfis ThalasemiaIndriyaniiIntandewataBelum ada peringkat
- Makalah ThalasemiaDokumen31 halamanMakalah ThalasemiaMasuhaidi MerlungBelum ada peringkat
- LAPORAN PENDAHULUAN Kejang DemamDokumen15 halamanLAPORAN PENDAHULUAN Kejang DemamanratBelum ada peringkat
- Kelainan Kongenital Pada Bayi Baru LahirDokumen6 halamanKelainan Kongenital Pada Bayi Baru LahirSimon Ganesya RahardjoBelum ada peringkat
- Periodik ParalisisDokumen15 halamanPeriodik ParalisisMuhammad Luthfi MunadhilBelum ada peringkat
- Anatomi Fisiologi DarahDokumen10 halamanAnatomi Fisiologi DarahFajar Joni Kurniawan100% (4)
- Klasifikasi ThalasemiaDokumen3 halamanKlasifikasi ThalasemiaNanda SachiBelum ada peringkat
- Manifestasi Klinis AutismeDokumen3 halamanManifestasi Klinis AutismeYohandhita DhitaBelum ada peringkat
- LeukimiaDokumen4 halamanLeukimiaGefaritza RabbaniBelum ada peringkat
- CtevDokumen6 halamanCtevSiti Nurjanah SeptianiBelum ada peringkat
- Demam ReumatikDokumen22 halamanDemam ReumatikRiskiyah MusyafirahBelum ada peringkat
- Kontraksi Otot (Kelompok 1)Dokumen19 halamanKontraksi Otot (Kelompok 1)Rina LestariBelum ada peringkat
- Kasus Penatalaksnaan Nutrisi Pada Pasien KritisDokumen3 halamanKasus Penatalaksnaan Nutrisi Pada Pasien KritisRachma Normalita DewiBelum ada peringkat
- Komplikasi HPPDokumen1 halamanKomplikasi HPPadilla kusumaBelum ada peringkat
- Pathway IMA by Diah WidiastitiDokumen3 halamanPathway IMA by Diah WidiastitiDiah WidiastitiBelum ada peringkat
- Transient Ischaemic Attack TIADokumen3 halamanTransient Ischaemic Attack TIARahmat FahrezaBelum ada peringkat
- Kolestasis JaundiceDokumen14 halamanKolestasis Jaundiceajipramana93Belum ada peringkat
- Askep Kritis StrokeDokumen13 halamanAskep Kritis StrokeMilu MardaniBelum ada peringkat
- Laporan Pendahuluan Kejang DemamDokumen17 halamanLaporan Pendahuluan Kejang Demamkevin martinBelum ada peringkat
- Hemofilia Pada AnakDokumen21 halamanHemofilia Pada AnakAri KurniawatiBelum ada peringkat
- Hemofilia Bu Maryatun (Kep Anak)Dokumen24 halamanHemofilia Bu Maryatun (Kep Anak)Ambika AnggardaniBelum ada peringkat
- Hemo FiliaDokumen9 halamanHemo FiliaNdok LhiaBelum ada peringkat
- Penanganan K3 Di Laboratorium KimiaDokumen8 halamanPenanganan K3 Di Laboratorium KimiaReza Sri MulfiaBelum ada peringkat
- Bank DarahDokumen3 halamanBank DarahReza Sri MulfiaBelum ada peringkat
- Analisis Data KualitatifDokumen13 halamanAnalisis Data Kualitatifikha yuliaBelum ada peringkat
- K3 Laboratorium BiokimiaDokumen14 halamanK3 Laboratorium BiokimiaReza Sri MulfiaBelum ada peringkat
- PLASMODIUM OVALE Kel.4Dokumen10 halamanPLASMODIUM OVALE Kel.4Reza Sri MulfiaBelum ada peringkat
- Makalah SPI Kelompok 12Dokumen20 halamanMakalah SPI Kelompok 12Reza Sri MulfiaBelum ada peringkat
- Makalah Askep HipertensiDokumen58 halamanMakalah Askep HipertensiReza Sri MulfiaBelum ada peringkat
- Terapi Modalitas Pada Sistem PernafasanDokumen3 halamanTerapi Modalitas Pada Sistem PernafasanReza Sri Mulfia AvgBelum ada peringkat
- Format Antenatal RevisiDokumen14 halamanFormat Antenatal RevisiReza Sri MulfiaBelum ada peringkat
- GTNDokumen6 halamanGTNcandra100% (1)
- Terapi Modalitas Pada Sistem PernafasanDokumen5 halamanTerapi Modalitas Pada Sistem PernafasanReza Sri Mulfia AvgBelum ada peringkat
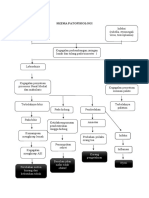
- Skema PatofisiologiDokumen1 halamanSkema PatofisiologiReza Sri MulfiaBelum ada peringkat
- Kep AjalDokumen8 halamanKep AjalReza Sri Mulfia AvgBelum ada peringkat
- Hubungan Kolesterol Dengan Penyakit JantungDokumen8 halamanHubungan Kolesterol Dengan Penyakit JantungReza Sri Mulfia AvgBelum ada peringkat
- Judul Jurnal KomDokumen4 halamanJudul Jurnal KomReza Sri MulfiaBelum ada peringkat
- PROSES KEPERAWATAN GERONTIK IniDokumen5 halamanPROSES KEPERAWATAN GERONTIK IniReza Sri Mulfia AvgBelum ada peringkat
- Analisis Jurnal ModalDokumen3 halamanAnalisis Jurnal ModallenafitriyaniBelum ada peringkat
- Berbagai Terapi Komplementer Berdasarkan JurnalDokumen3 halamanBerbagai Terapi Komplementer Berdasarkan JurnalReza Sri Mulfia AvgBelum ada peringkat
- Community Mental Health NursingDokumen12 halamanCommunity Mental Health NursingReza Sri MulfiaBelum ada peringkat
- Keperawatan Jiwa Terkait Resiko Prilaku Kekerasan Dan Resiko Bunuh DiriDokumen15 halamanKeperawatan Jiwa Terkait Resiko Prilaku Kekerasan Dan Resiko Bunuh DiriReza Sri Mulfia AvgBelum ada peringkat
- Bencana Pada Lanjut UsiaDokumen11 halamanBencana Pada Lanjut UsiaAmaliaBelum ada peringkat
- Community Mental Health NursingDokumen12 halamanCommunity Mental Health NursingReza Sri MulfiaBelum ada peringkat
- Bab 2 HomecareDokumen24 halamanBab 2 HomecareReza Sri Mulfia AvgBelum ada peringkat
- Bab 2 HomecareDokumen6 halamanBab 2 HomecareReza Sri Mulfia AvgBelum ada peringkat
- Penyuluhan KGDDokumen2 halamanPenyuluhan KGDReza Sri MulfiaBelum ada peringkat
- Kata PengantarDokumen3 halamanKata PengantarReza Sri MulfiaBelum ada peringkat
- Tumbuh Kembang NeonatusDokumen26 halamanTumbuh Kembang NeonatusReza Sri MulfiaBelum ada peringkat
- Semester 4 Maternitas Kelompok 2Dokumen14 halamanSemester 4 Maternitas Kelompok 2Reza Sri MulfiaBelum ada peringkat