Anda mungkin juga menyukai
- PolarimeterDokumen11 halamanPolarimeterVanzoell95Belum ada peringkat
- AAS SpektrofotometriDokumen11 halamanAAS SpektrofotometriWulannian AnwariBelum ada peringkat
- KATABOLISME PROTEINDokumen3 halamanKATABOLISME PROTEINaisyah fajrinBelum ada peringkat
- ANALISIS KADAR LOGAM DENGAN FOTOMETER NYALADokumen12 halamanANALISIS KADAR LOGAM DENGAN FOTOMETER NYALAAristy MirandaBelum ada peringkat
- Laporan FurnaceDokumen9 halamanLaporan FurnaceMuhammad Nur ArifinBelum ada peringkat
- SPEKTRODokumen11 halamanSPEKTROrina febrinaBelum ada peringkat
- PolarimeterDokumen8 halamanPolarimeterAl YudhaBelum ada peringkat
- Spektro MCDokumen10 halamanSpektro MCAndi Nadya SorayaBelum ada peringkat
- Kromatografi PlanarDokumen5 halamanKromatografi PlanardinipertamaBelum ada peringkat
- Revisi Makalah Waterbath - Kelompok 7 - Pbio ADokumen13 halamanRevisi Makalah Waterbath - Kelompok 7 - Pbio AAsti ApriliaBelum ada peringkat
- POTENSIOMETRIDokumen6 halamanPOTENSIOMETRIGitha Jha DechBelum ada peringkat
- Analisis Sampel Tembaga dengan UV-VISDokumen22 halamanAnalisis Sampel Tembaga dengan UV-VISAnnisaBelum ada peringkat
- PH MeterDokumen13 halamanPH Meterasa attalya amaziaBelum ada peringkat
- Laporan AasDokumen16 halamanLaporan AasNuzul Al-QoriBelum ada peringkat
- Spektrofotometer Adalah Alat Untuk Mengukur Transmitan Atau Absorban Suatu Sampel Sebagai Fungsi Panjang GelombangDokumen1 halamanSpektrofotometer Adalah Alat Untuk Mengukur Transmitan Atau Absorban Suatu Sampel Sebagai Fungsi Panjang Gelombangrizaldi rahmatullahBelum ada peringkat
- Laporan Praktikum ConveyorDokumen9 halamanLaporan Praktikum ConveyorcalvinBelum ada peringkat
- KROMATOGRAFI KERTAS PMBDokumen11 halamanKROMATOGRAFI KERTAS PMBNovVie VietTha Sccor IIBelum ada peringkat
- YakultDokumen20 halamanYakultFirdayanti AdamBelum ada peringkat
- Membran SelDokumen4 halamanMembran SelFase Husnu LestariBelum ada peringkat
- Spektrofotometri UV VISDokumen7 halamanSpektrofotometri UV VISardiansyahds23Belum ada peringkat
- GC-MS TEKNIK ANALISISDokumen5 halamanGC-MS TEKNIK ANALISISAndike Rahma NandaBelum ada peringkat
- Makalah Kelompok 2 Pipet 03tkme0001Dokumen20 halamanMakalah Kelompok 2 Pipet 03tkme0001Vhannesa Nur RahmaBelum ada peringkat
- TURBIDIMETERDokumen13 halamanTURBIDIMETERSurya AdeliaBelum ada peringkat
- Makalah Kelompok 4 Siklus Sel Dan Kanker PDFDokumen55 halamanMakalah Kelompok 4 Siklus Sel Dan Kanker PDFcheril saseaBelum ada peringkat
- Menerapkan Prinsip Perawatan Alat-Alat Di Laboratorium KimiaDokumen11 halamanMenerapkan Prinsip Perawatan Alat-Alat Di Laboratorium Kimianur fauzia100% (1)
- KTI TUGAS AKHIR AGUNGS BERSATU Lampiran & MalBook PDFDokumen66 halamanKTI TUGAS AKHIR AGUNGS BERSATU Lampiran & MalBook PDFIsma BondolBelum ada peringkat
- Isolasi Protein JaringanDokumen9 halamanIsolasi Protein JaringanNuri OktarinaBelum ada peringkat
- Makalah Alat CentrifugeDokumen18 halamanMakalah Alat CentrifugeNarty pratamaBelum ada peringkat
- Laporan Praktikum AASDokumen10 halamanLaporan Praktikum AASEki Ruskartina100% (1)
- Lab KeselamatanDokumen11 halamanLab KeselamatanYohan AnsanayBelum ada peringkat
- Laporan Praktikum PENGENALAN GCDokumen18 halamanLaporan Praktikum PENGENALAN GCRini RositaBelum ada peringkat
- Elektroforesis KertasDokumen8 halamanElektroforesis KertasFood Tech 2022Belum ada peringkat
- Eltia Putri - A - Neraca AnalitikDokumen5 halamanEltia Putri - A - Neraca AnalitikEltia PutriBelum ada peringkat
- Analisis Metilen Biru dengan SpektrofotometriDokumen20 halamanAnalisis Metilen Biru dengan SpektrofotometriReynand ThoriqBelum ada peringkat
- Spektrofotometri Serapan AtomDokumen22 halamanSpektrofotometri Serapan AtomMario HuangBelum ada peringkat
- Makalah K3 Kel.2Dokumen33 halamanMakalah K3 Kel.2Jeni SumuriBelum ada peringkat
- Laprak Pembuatan Pati Jagung Dan Susu JagungDokumen25 halamanLaprak Pembuatan Pati Jagung Dan Susu JagungMustikaNindiyaBelum ada peringkat
- Laporan Uji Nyala Dan Indetifikasi GugusDokumen18 halamanLaporan Uji Nyala Dan Indetifikasi GugusTeriany WidjayaBelum ada peringkat
- Laporan Aas PBDokumen18 halamanLaporan Aas PByunitiaBelum ada peringkat
- Job Safety AnalysisDokumen24 halamanJob Safety AnalysisMUHAMMAD HAFIDZBelum ada peringkat
- LAPORAN KLP 1 DENSITOMETERDokumen5 halamanLAPORAN KLP 1 DENSITOMETERHasri NinisBelum ada peringkat
- SOP AAS & PotensiometerDokumen21 halamanSOP AAS & PotensiometerBerta Yuda Sisilia Putri100% (2)
- Spektrofotometri Penjelasan Istilah & Fungsi Kurva KalibrasiDokumen1 halamanSpektrofotometri Penjelasan Istilah & Fungsi Kurva KalibrasiistimahillahBelum ada peringkat
- Gravimetri Kadar AirDokumen4 halamanGravimetri Kadar AirDini FisadiahBelum ada peringkat
- Laporan Praktikum KalibrasiDokumen7 halamanLaporan Praktikum KalibrasiNurliza AprilianiBelum ada peringkat
- X Penetapan Kadar Kalium Dan Aluminium Menggunakan AASDokumen22 halamanX Penetapan Kadar Kalium Dan Aluminium Menggunakan AAShafiz khanBelum ada peringkat
- OPTIMASI DRYING OVENDokumen25 halamanOPTIMASI DRYING OVENNeo BagusBelum ada peringkat
- Analisis Kalsium AirDokumen22 halamanAnalisis Kalsium AirEphy AprianyBelum ada peringkat
- Laporan AasDokumen26 halamanLaporan AasBenni Pranatal Cristian Sirait100% (2)
- Laporan Praktikum Instrumen Hot Plate ZalwaDokumen5 halamanLaporan Praktikum Instrumen Hot Plate ZalwaDawa AeginaBelum ada peringkat
- Tugas SpektrofotometerDokumen14 halamanTugas SpektrofotometerDikin DBelum ada peringkat
- KALIBRASI SPEKTRODokumen6 halamanKALIBRASI SPEKTROrisha octaviaBelum ada peringkat
- Flame FotometerDokumen12 halamanFlame FotometerJ Fadli100% (1)
- KADAR GULADokumen6 halamanKADAR GULAArian Rizki WardanaBelum ada peringkat
- LAPORAN PRAKTIKUM TPP 2 Oven MicrowaveDokumen26 halamanLAPORAN PRAKTIKUM TPP 2 Oven MicrowaveFadhilAlfiyantoQ100% (1)
- CentrifugeDokumen9 halamanCentrifugeAmiraBelum ada peringkat
- Spektrometri Serapan Atom (Ssa)Dokumen11 halamanSpektrometri Serapan Atom (Ssa)Annisa Soraya UlfaBelum ada peringkat
- Laporan Praktikum Anis AASDokumen23 halamanLaporan Praktikum Anis AASDeisy Indayanti89% (9)
- Spektrofotometri Serapan Atom 1Dokumen15 halamanSpektrofotometri Serapan Atom 1nananananaBelum ada peringkat
- Laporan Praktikum Anis AASDokumen23 halamanLaporan Praktikum Anis AASandika fikramBelum ada peringkat
- KSP Kim3 2 PDFDokumen2 halamanKSP Kim3 2 PDFFitri AlawiyahBelum ada peringkat
- Jadwal PantukhirDokumen1 halamanJadwal Pantukhirangga sahputra siraitBelum ada peringkat
- Perka BAPETEN No 1 TAHUN 2010 Tentang KESIAPSIAGAAN DAN PENANGGULANGAN KEDARURATAN NUKLIRDokumen56 halamanPerka BAPETEN No 1 TAHUN 2010 Tentang KESIAPSIAGAAN DAN PENANGGULANGAN KEDARURATAN NUKLIRnirwonommBelum ada peringkat
- 2 PDFDokumen170 halaman2 PDFFitri AlawiyahBelum ada peringkat
- Booklet Beasiswa Reguler Tahun 2019Dokumen12 halamanBooklet Beasiswa Reguler Tahun 2019eka kartikaBelum ada peringkat
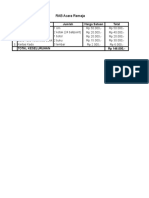
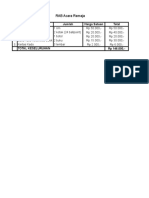
- RAB Acara Remaj-WPS OfficeDokumen1 halamanRAB Acara Remaj-WPS OfficeFitri AlawiyahBelum ada peringkat
- PK-149 Tugas Individu Institutional Visit BUMNDokumen1 halamanPK-149 Tugas Individu Institutional Visit BUMNFitri AlawiyahBelum ada peringkat
- RAB Acara RemajaDokumen1 halamanRAB Acara RemajaFitri AlawiyahBelum ada peringkat
- Tugas Per-UU - Fitri Alawiyah - Teknokimia Nuklir 2014Dokumen5 halamanTugas Per-UU - Fitri Alawiyah - Teknokimia Nuklir 2014Fitri AlawiyahBelum ada peringkat
- Dapus ToksikDokumen1 halamanDapus ToksikFitri AlawiyahBelum ada peringkat
- Etika Profesi (Standar-Standar)Dokumen2 halamanEtika Profesi (Standar-Standar)Fitri AlawiyahBelum ada peringkat
- Tugas RekKimDokumen15 halamanTugas RekKimFitri AlawiyahBelum ada peringkat
- Fitri Alawiyah - 011400381 - Teknokimia Nuklir 2014Dokumen4 halamanFitri Alawiyah - 011400381 - Teknokimia Nuklir 2014Fitri AlawiyahBelum ada peringkat
- Etika Profesi (Standar-Standar)Dokumen2 halamanEtika Profesi (Standar-Standar)Fitri AlawiyahBelum ada peringkat
- Tugas Per-UU - Fitri Alawiyah - Teknokimia Nuklir 2014Dokumen5 halamanTugas Per-UU - Fitri Alawiyah - Teknokimia Nuklir 2014Fitri AlawiyahBelum ada peringkat
- Panduan Menggunakan Whaff RewardDokumen4 halamanPanduan Menggunakan Whaff RewardFitri AlawiyahBelum ada peringkat
- BOD, COD & TSSDokumen2 halamanBOD, COD & TSSFitri Alawiyah100% (1)
- Makalah Operasi Teknik Kimia IIDokumen17 halamanMakalah Operasi Teknik Kimia IIFitri AlawiyahBelum ada peringkat
- MriDokumen5 halamanMriFitri AlawiyahBelum ada peringkat
- Sosiologi Agama Agama Dan Konflik Sosial Hanni Khotimah PDFDokumen12 halamanSosiologi Agama Agama Dan Konflik Sosial Hanni Khotimah PDFFitri AlawiyahBelum ada peringkat
- Ilmu Bahan (Kelompok 3)Dokumen38 halamanIlmu Bahan (Kelompok 3)Fitri AlawiyahBelum ada peringkat
- Tugas Pre-Test Pak HariesDokumen17 halamanTugas Pre-Test Pak HariesFitri AlawiyahBelum ada peringkat
- Penentuan Bilangan AsamDokumen1 halamanPenentuan Bilangan AsamFitri AlawiyahBelum ada peringkat
- Makalah Instrumentasi Medik 1Dokumen22 halamanMakalah Instrumentasi Medik 1Fitri AlawiyahBelum ada peringkat
- Tugas PKBNDokumen1 halamanTugas PKBNFitri AlawiyahBelum ada peringkat
- BHN STRKDokumen23 halamanBHN STRKFitri AlawiyahBelum ada peringkat
- Ringkasan Pengelolaan LimbahDokumen7 halamanRingkasan Pengelolaan LimbahFitri AlawiyahBelum ada peringkat
- Kelompok Praktikum PemrogramanDokumen1 halamanKelompok Praktikum PemrogramanFitri AlawiyahBelum ada peringkat
- PKBNDokumen3 halamanPKBNFitri AlawiyahBelum ada peringkat
- Tugas Bu Maria Di BukuDokumen6 halamanTugas Bu Maria Di BukuFitri AlawiyahBelum ada peringkat