Anda mungkin juga menyukai
- Infus & Transfusi TukarDokumen53 halamanInfus & Transfusi TukarvanthygBelum ada peringkat
- Konsep Dasar Transfusi DarahDokumen33 halamanKonsep Dasar Transfusi DarahRizqi KhusnulBelum ada peringkat
- Asuhan Keperawatan HemofiliaDokumen30 halamanAsuhan Keperawatan HemofiliaRumah Daster Mantangai100% (1)
- THALASEMIA PADA ANAKDokumen16 halamanTHALASEMIA PADA ANAKFebrio RioBelum ada peringkat
- LP Thalasemia AnakDokumen16 halamanLP Thalasemia AnakYuliantiBelum ada peringkat
- LP Anak Thalasemia Ruslandi - SKEPDokumen22 halamanLP Anak Thalasemia Ruslandi - SKEPSalman FirmansyahBelum ada peringkat
- Jurnal PicoDokumen7 halamanJurnal PicoArisa ViraBelum ada peringkat
- LP CA ColorektalDokumen18 halamanLP CA Colorektaledi sambasBelum ada peringkat
- SEPSISDokumen23 halamanSEPSISBee MullanaBelum ada peringkat
- GANGGUAN TIDUR LANSIADokumen11 halamanGANGGUAN TIDUR LANSIADadahlia LaraBelum ada peringkat
- LP Thalasemia Tahun TerbaruDokumen19 halamanLP Thalasemia Tahun TerbaruWahyu Eka WulandariBelum ada peringkat
- Patoflow HemofiliaDokumen3 halamanPatoflow HemofiliaRosa hartiniBelum ada peringkat
- LP ThalasemiaDokumen12 halamanLP ThalasemiaTri Nur JayantiBelum ada peringkat
- Analisa Tindakan TRANSFUSI DARAHDokumen5 halamanAnalisa Tindakan TRANSFUSI DARAHRohani sakimanBelum ada peringkat
- Woc Kelompok 6Dokumen1 halamanWoc Kelompok 6Nur ElaeniBelum ada peringkat
- Poa Keperawatan KomunitasDokumen4 halamanPoa Keperawatan KomunitasYusrilBelum ada peringkat
- TrendelenburgDokumen23 halamanTrendelenburgFauzi IskandarBelum ada peringkat
- Reaksi 1.2Dokumen19 halamanReaksi 1.2Intan SariBelum ada peringkat
- Asuhan Keperawatan Pada Anak Dengan ITPDokumen13 halamanAsuhan Keperawatan Pada Anak Dengan ITPLyra Make UpBelum ada peringkat
- PatofisiologiDokumen2 halamanPatofisiologirima ayu fitriana100% (1)
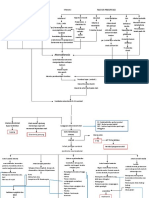
- Pathway ALLDokumen1 halamanPathway ALLAyu IndriBelum ada peringkat
- Konsep Map DHFDokumen3 halamanKonsep Map DHFigd100% (1)
- LP MDS KMB 1Dokumen12 halamanLP MDS KMB 1qurrotul ayunBelum ada peringkat
- Mode VentilatorDokumen3 halamanMode VentilatorJelisaaBelum ada peringkat
- HIPERBILIRUBINEMIADokumen21 halamanHIPERBILIRUBINEMIAMadona RustiainiBelum ada peringkat
- ALL Asuhan KeperawatanDokumen14 halamanALL Asuhan KeperawatanFitri UtamiBelum ada peringkat
- Laporan Pendahuluan MyelofibrosisDokumen18 halamanLaporan Pendahuluan MyelofibrosisliayusfianaBelum ada peringkat
- Makalah Gangguan Sistem Hematologi Askep Anemia Defisiensi BesiDokumen23 halamanMakalah Gangguan Sistem Hematologi Askep Anemia Defisiensi BesiPhiedta Purnamasari RahmadhaniBelum ada peringkat
- Perawatan Anak Sakit Di RumahDokumen2 halamanPerawatan Anak Sakit Di RumahRamadhan GhaffarBelum ada peringkat
- MAKALAH PEMASANGAN DESFERALDokumen13 halamanMAKALAH PEMASANGAN DESFERALRatihBelum ada peringkat
- Kelompok 8 - ThalasemiaDokumen21 halamanKelompok 8 - ThalasemiaVidya AnandaBelum ada peringkat
- LP ThalasemiaDokumen7 halamanLP ThalasemiaHeryzanurBelum ada peringkat
- ASKEP LeukimiaDokumen14 halamanASKEP LeukimiaARIVANSELBelum ada peringkat
- Konsep Dasar Penyakit TalasemiaDokumen23 halamanKonsep Dasar Penyakit TalasemiariskaBelum ada peringkat
- Tumbuhan Obat Suku RejangDokumen86 halamanTumbuhan Obat Suku RejangOka Iramda SaputraBelum ada peringkat
- LP 4 Komunikasi Efektif SBAR Atau ISBAR-1Dokumen4 halamanLP 4 Komunikasi Efektif SBAR Atau ISBAR-1Muhammad HarimansyabBelum ada peringkat
- MCP SYOK HIPOVOLEMIK (Pingky Anggraeny) - DikonversiDokumen20 halamanMCP SYOK HIPOVOLEMIK (Pingky Anggraeny) - DikonversiPingky AnggraenyBelum ada peringkat
- Askep Thalasemia Pada AnakDokumen18 halamanAskep Thalasemia Pada AnakRezki wahyuniBelum ada peringkat
- Askep Pada AneurysmaDokumen29 halamanAskep Pada AneurysmaFransiscaapBelum ada peringkat
- Definisi Dan Tujuan Initial AssesmentDokumen1 halamanDefinisi Dan Tujuan Initial AssesmentNathalia Rose Fransisca KarmaBelum ada peringkat
- Asuhan Keperawatan Katup JantungDokumen24 halamanAsuhan Keperawatan Katup JantungwidyaBelum ada peringkat
- Diaper RashDokumen7 halamanDiaper RashMaghfirahEkasariLaitjinaraBelum ada peringkat
- Referat Transfusi Tukar Dan FototerapiDokumen20 halamanReferat Transfusi Tukar Dan FototerapiWahid KasturyBelum ada peringkat
- Pathway NHS Frendhy de Fretes. Stik Stella Maris MakassarDokumen3 halamanPathway NHS Frendhy de Fretes. Stik Stella Maris MakassarfrendhyBelum ada peringkat
- ASDDokumen1 halamanASDmelinaBelum ada peringkat
- Patofisiologi Syok HipovolemikDokumen3 halamanPatofisiologi Syok HipovolemikYuniBelum ada peringkat
- LK Ca VulvaDokumen13 halamanLK Ca VulvaDennyDwiPinasti100% (1)
- TALASEMIADokumen23 halamanTALASEMIASri Mahtufa RiskiBelum ada peringkat
- Pathway DM FiksDokumen2 halamanPathway DM FiksputuwidiaBelum ada peringkat
- Transfusi Darah Pada Bayi & AnakDokumen27 halamanTransfusi Darah Pada Bayi & AnakHameldo Andika PattinasaranyBelum ada peringkat
- Thalasemia pada AnakDokumen4 halamanThalasemia pada AnakElla Effendi22Belum ada peringkat
- Syok Hipovolemik: Penyebab, Gejala, dan PenanganannyaDokumen17 halamanSyok Hipovolemik: Penyebab, Gejala, dan PenanganannyaAyu Nur HeafenBelum ada peringkat
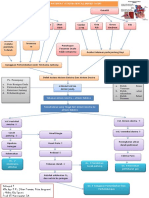
- SEOOptimalJudulKurang40KarakterUntukDokumenWOCDokumen11 halamanSEOOptimalJudulKurang40KarakterUntukDokumenWOCPuti FatimahBelum ada peringkat
- VSDDokumen22 halamanVSDSafitri UlandaryBelum ada peringkat
- KANKER PROSTATDokumen13 halamanKANKER PROSTATHafaf RaufBelum ada peringkat
- Woc-Nstemi PDFDokumen1 halamanWoc-Nstemi PDFSannty DjampangBelum ada peringkat
- LP MDSDokumen13 halamanLP MDSsulchafitriyaBelum ada peringkat
- Klasifikasi ThalasemiaDokumen3 halamanKlasifikasi ThalasemiaNanda SachiBelum ada peringkat
- Kelompok 5 - Makalah Penugasan 1 - SpritualitasCBDokumen12 halamanKelompok 5 - Makalah Penugasan 1 - SpritualitasCBmarchelinaeva223Belum ada peringkat
- Laporan Pendahuluan Talasemia (Octavia Husna N.A 17613084)Dokumen25 halamanLaporan Pendahuluan Talasemia (Octavia Husna N.A 17613084)OvieavioBelum ada peringkat
- Presentasi Thalasemia AnakDokumen24 halamanPresentasi Thalasemia Anakkeuangan medisaBelum ada peringkat
- MetabolismeDokumen14 halamanMetabolismeArisa ViraBelum ada peringkat
- RIWAYAT KESEHATANDokumen9 halamanRIWAYAT KESEHATANArisa ViraBelum ada peringkat
- Asuhan Keperawatan DMDokumen14 halamanAsuhan Keperawatan DMArisa ViraBelum ada peringkat
- CONTOH ABSTRAK PenelitianDokumen4 halamanCONTOH ABSTRAK PenelitianArisa ViraBelum ada peringkat
- TBCDokumen8 halamanTBCArisa ViraBelum ada peringkat
- Askep Curret AnisaDokumen26 halamanAskep Curret AnisaArisa ViraBelum ada peringkat
- Arisa Vira - Askep Kritis KadDokumen29 halamanArisa Vira - Askep Kritis KadArisa ViraBelum ada peringkat
- PANUM PPT Askep GadarDokumen13 halamanPANUM PPT Askep GadarArisa ViraBelum ada peringkat
- Materi Pendidikan Kesehatan Rinitis Medikamentosa Akibat Penggunaan Dekongestan TopikalDokumen8 halamanMateri Pendidikan Kesehatan Rinitis Medikamentosa Akibat Penggunaan Dekongestan TopikalArisa ViraBelum ada peringkat
- Pengkajian AnakDokumen3 halamanPengkajian AnakArisa ViraBelum ada peringkat
- SOP Postpartum - Kel 3Dokumen7 halamanSOP Postpartum - Kel 3Arisa ViraBelum ada peringkat
- Arisa Vira Oktafiani - Kep - Gadar - Trend & Issue Covid - Bu WidiDokumen5 halamanArisa Vira Oktafiani - Kep - Gadar - Trend & Issue Covid - Bu WidiArisa ViraBelum ada peringkat
- Play Isolasi SosialDokumen5 halamanPlay Isolasi SosialArisa ViraBelum ada peringkat
- Sap Gizi YulindaDokumen11 halamanSap Gizi YulindaArisa ViraBelum ada peringkat
- SETTING KAMAR BEDAHDokumen20 halamanSETTING KAMAR BEDAHArisa ViraBelum ada peringkat
- Askep Kelompok Anak KDMDokumen17 halamanAskep Kelompok Anak KDMArisa ViraBelum ada peringkat
- Askep Bayi HiperbilirubinemiaDokumen45 halamanAskep Bayi HiperbilirubinemiaHelgi Prasetya NugrahaBelum ada peringkat
- Pemeriksaan Head To ToeDokumen1 halamanPemeriksaan Head To ToeArisa ViraBelum ada peringkat
- KEPERAWATAN GADARDokumen14 halamanKEPERAWATAN GADARArisa ViraBelum ada peringkat
- Kasus Encepalitis - TugasDokumen8 halamanKasus Encepalitis - TugasArisa ViraBelum ada peringkat
- Kel 6 KeluargaDokumen23 halamanKel 6 KeluargaArisa ViraBelum ada peringkat
- Arisa Laporan DMDokumen22 halamanArisa Laporan DMArisa ViraBelum ada peringkat
- Resume Teori Model Keperawatan AnakDokumen6 halamanResume Teori Model Keperawatan AnakArisa ViraBelum ada peringkat
- Askep-Hiperbilirubin PDFDokumen37 halamanAskep-Hiperbilirubin PDFLa HajirinBelum ada peringkat
- Format Askep AnakDokumen8 halamanFormat Askep AnakArisa ViraBelum ada peringkat
- Kelompok 7 Intrapartum Bu IndahDokumen23 halamanKelompok 7 Intrapartum Bu IndahArisa ViraBelum ada peringkat
- Contoh Jadwal Kepala TimDokumen2 halamanContoh Jadwal Kepala TimArisa ViraBelum ada peringkat
- Contoh Jadwal Kepala TimDokumen2 halamanContoh Jadwal Kepala TimArisa ViraBelum ada peringkat